Anonymity networks are specialized networks designed to provide users with privacy and anonymity while they browse the internet or communicate online. These networks anonymize user data by routing internet traffic through a series of intermediate nodes or servers, making it difficult for outside observers to trace the origin of the data or identify the user. Some key characteristics and features of anonymity networks include: 1. **Privacy Protection**: They mask the user’s IP address, allowing them to browse the internet without revealing their true identity or location.
Ransomware is a type of malicious software (malware) that encrypts a victim's files or locks them out of their systems, making the data inaccessible. The attacker then demands a ransom payment, typically in cryptocurrency, to provide the decryption key or restore access to the compromised systems. Ransomware attacks can target individuals, businesses, and even government institutions. The impact of such attacks can be severe, leading to data loss, financial losses, operational disruption, and reputational damage.
A pseudonym is a fictitious name used by an author, artist, or individual instead of their real name. This practice is often employed for various reasons, such as to maintain anonymity, create a distinct persona, avoid legal issues, or separate different genres of work. For example, the famous British author Samuel Langhorne Clemens wrote under the pseudonym Mark Twain.
Publicly Verifiable Secret Sharing (PVSS) is a cryptographic scheme that allows a secret to be shared among several participants in such a way that the secret can be reconstructed only by a designated group of participants, while also allowing anyone to verify the correctness of the share distributions and the reconstruction of the secret.
OpenPuff is an open-source steganography tool that allows users to conceal data within various types of files, such as images, audio, and video. It utilizes advanced techniques to hide information in a way that is not easily detectable, making it a useful tool for those interested in privacy and secure communication. OpenPuff supports multiple levels of encryption and can hide large amounts of data within relatively small cover files.
Shuffle play is a feature commonly found in music and video streaming services that enables users to listen to or watch content in a random order, rather than in a predetermined sequence. When shuffle play is activated, the platform randomly selects songs, videos, or other media, creating a different listening or viewing experience each time. This is particularly useful for users who want to discover new music or content from a playlist or library without having to follow a specific order.
Language interpretation is the process of converting spoken or signed communication from one language to another in real-time. It is commonly used in settings such as conferences, meetings, legal proceedings, medical appointments, and other situations where effective communication between speakers of different languages is necessary. There are different modes of interpretation, including: 1. **Simultaneous Interpretation**: The interpreter translates the speaker's message into the target language in real-time, often using headphones and microphones.
The Global Consciousness Project (GCP) is a research initiative that aims to investigate the potential correlations between global events and collective human consciousness. It was initiated in 1998 by physicist Roger D. Nelson at Princeton University and uses a network of random number generators (RNGs) around the world to collect data.
Sortition is a method of selecting individuals for positions of authority or decision-making through a random selection process, rather than through elections or appointments. It is often associated with ancient Athenian democracy, where citizens were chosen by lot to fill various public offices and to serve on juries, reflecting the belief that all citizens should have an equal chance to participate in governance. The process of sortition is based on the idea that random selection can reduce bias and ensure a more representative sample of the population.
COBUILD (Collins Birmingham University International Language Database) is a specialized linguistic project that originated in the early 1980s. It focuses on creating dictionaries and language resources based on real-world examples of how English is used in context. The COBUILD project was initiated by a team at the University of Birmingham in the UK and is known for its innovative approach to dictionary-making. One of COBUILD's most prominent outputs is the "COBUILD English Dictionary," which was first published in 1987.
In linguistics, "error" refers to a deviation from the norms of a language, which can occur at various levels—phonetic, morphological, syntactic, semantic, or pragmatic. Errors typically arise when speakers or writers do not adhere to the grammatical rules, vocabulary, or pronunciation standards of a language. These deviations can occur in both first language (L1) and second language (L2) contexts.
"Danda" can refer to several things depending on the context: 1. **Cultural/Spiritual Context**: In some South Asian traditions, particularly in Hinduism and Buddhism, "Danda" refers to a staff or rod, often symbolizing authority or power. It can be associated with various deities and is sometimes used in rituals.
Boris Trakhtenbrot was a prominent Soviet mathematician known for his contributions to various fields, including mathematical logic and computer science. He was particularly noted for his work on the theory of algorithms and the foundations of mathematics. One of his significant contributions is the development of algorithms for decision problems and his work on the theory of automata.
Moksha is a concept in Indian philosophy and religions, particularly in Hinduism, Buddhism, Jainism, and Sikhism. It refers to the liberation or release from the cycle of birth, death, and rebirth (samsara). Achieving moksha is considered the ultimate goal of life in these traditions, representing spiritual freedom, enlightenment, and the realization of one's true self or unity with the divine.
Ivan Stojmenović is a notable figure known primarily for his contributions to the fields of computer science and engineering, particularly in areas related to networking, algorithms, and parallel processing. His research often involves topics such as wireless networks, mobile computing, and algorithm design. He is also recognized for his academic work, including publications in scientific journals and conferences.
As of my last knowledge update in October 2023, there is no widely known figure named Vladimir Dragović in public discourse, literature, science, or popular culture. It's possible that he could be a less-known individual in a specific field or a fictional character.
Rudolf Bauer was an Austrian politician, best known for his role as a member of the Austrian Parliament. He belonged to the Social Democratic Party (SPÖ) and was involved in various legislative efforts during his political career. Details about his specific contributions and political ideology would need to be researched further, as contemporary information may be limited.
Emanuel Derman is a prominent figure in the fields of finance and quantitative modeling. He is best known for his contributions to financial engineering and for his work on options pricing models. Derman is often associated with the development of the Black-Derman-Toy model, which is an interest rate model used in the pricing of options and other financial derivatives. In addition to his work in finance, Derman has a background in physics, having earned a Ph.D. in theoretical physics from Columbia University.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.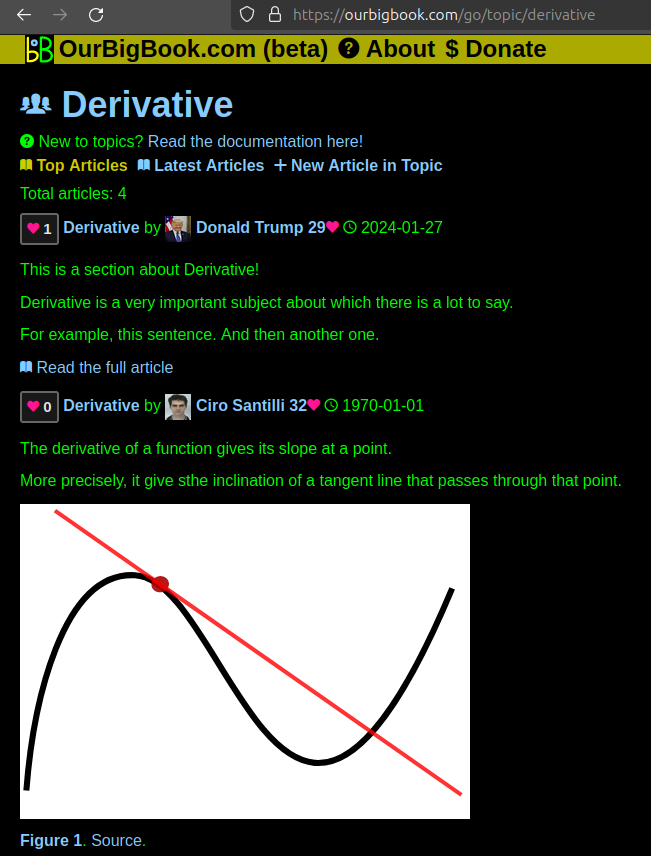
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.
Figure 4. Visual Studio Code extension tree navigation.
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 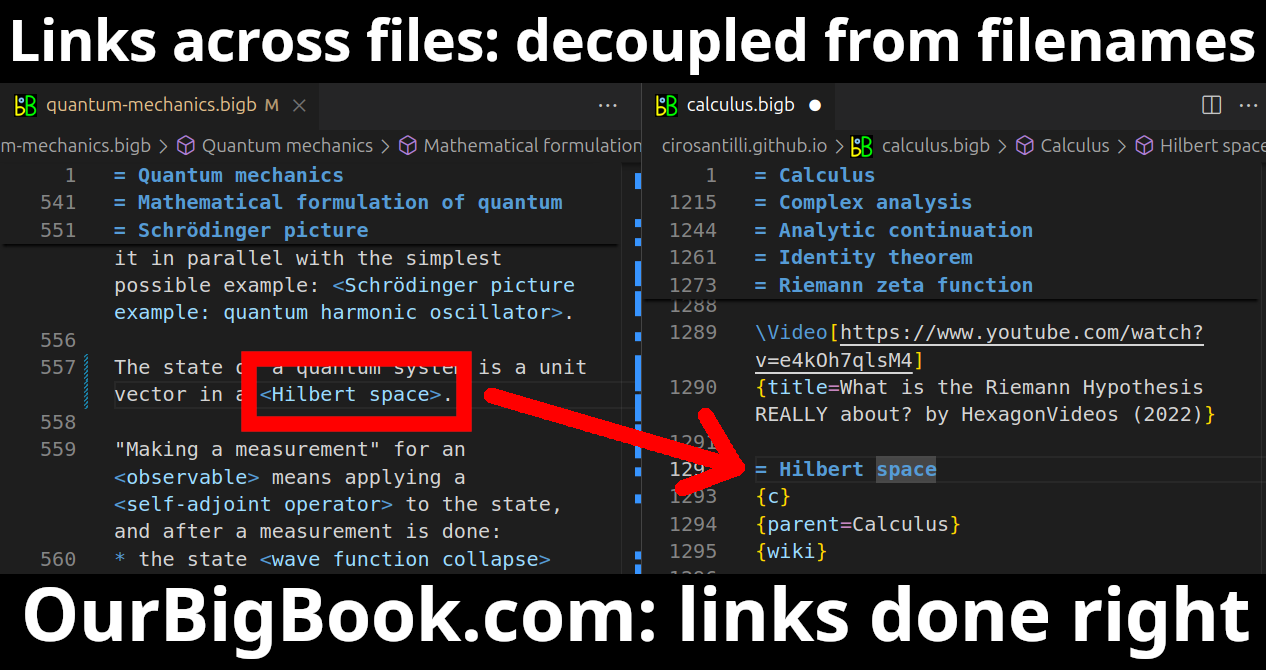
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact