Macdonald identities are a set of identities in the theory of symmetric functions, named after I.G. Macdonald. These identities relate certain algebraic structures known as symmetric functions, particularly the Macdonald polynomials, to various combinatorial objects. The identities typically express symmetric polynomials, which can be thought of as generating functions for certain combinatorial objects, in terms of other symmetric polynomials.
The quintuple product identity is a mathematical identity related to the theory of partitions and q-series, often involving generating functions in combinatorial contexts. It is a specific case of the more general product identities that arise in the theory of modular forms and q-series.
Cksum, or "checksum," is a utility commonly used in computing and telecommunications to verify the integrity of data. A checksum is a value that is calculated from a data set (like a file or a block of memory) to help ensure that the data has not been altered or corrupted during transmission or storage. When data is transmitted or saved, a checksum is generated based on the contents of the data.
Vaughan's identity is an important result in analytic number theory, particularly in the context of additive number theory and the study of sums of arithmetic functions. The identity provides a way to express the sum of a function over a set of integers in terms of more manageable sums and is often used in the context of problems involving the distribution of prime numbers.
Homogeneous polynomials are a special class of polynomials that have the property that all their terms have the same total degree. In mathematical terms, a polynomial \( P(x_1, x_2, \ldots, x_n) \) is considered homogeneous of degree \( d \) if every term in the polynomial is of degree \( d \).
Orthogonal polynomials are a class of polynomials that satisfy specific orthogonality conditions with respect to a given weight function over a certain interval.
The Lagrange polynomial is a form of polynomial interpolation used to find a polynomial that passes through a given set of points.
The term "Neumann polynomial" is not widely recognized in mathematical literature. However, it seems you might be referring to the "Neumann series" or the "Neumann problem" in the context of mathematics, particularly in functional analysis or differential equations. 1. **Neumann Series**: This refers to a specific type of series related to the inverses of operators.
Polynomial interpolation is a mathematical method used to estimate or approximate a polynomial function that passes through a given set of data points.
A Q-difference polynomial is an extension of the classical notion of polynomials in the context of difference equations and q-calculus. It is primarily used in the field of quantum calculus, where the concept of q-analogues is prevalent. In a basic sense, a Q-difference polynomial can be viewed as a polynomial where the variable \( x \) is replaced by \( q^x \), where \( q \) is a fixed non-zero complex number (often assumed to be non-negative).
A trigonometric polynomial is a mathematical expression that is composed of a finite sum of sine and cosine functions.
Kostka numbers, denoted as \( K_{n, \lambda} \), arise in combinatorial representation theory and algebraic geometry. They count the number of ways to arrange a certain type of tableau (specifically, standard Young tableaux) corresponding to partitions and is related to the representation theory of the symmetric group.
The Koecher–Vinberg theorem is a result in the field of arithmetic geometry, specifically concerning the structure of certain types of algebraic varieties. This theorem is particularly relevant in the study of symmetric spaces and the theory of quadratic forms. In broad terms, the Koecher–Vinberg theorem addresses the behavior of closed cones in the context of the theory of quadratic forms, stating conditions under which certain cones can be regarded as "nice" with respect to their arithmetic and geometric properties.
Niven's theorem is a result in number theory that concerns the rationality of certain integrals. Specifically, it states that if \( a \) is a positive integer, then the integral \[ \int_0^1 x^a (1 - x)^a \, dx \] is a rational number and can be expressed in terms of the binomial coefficient.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.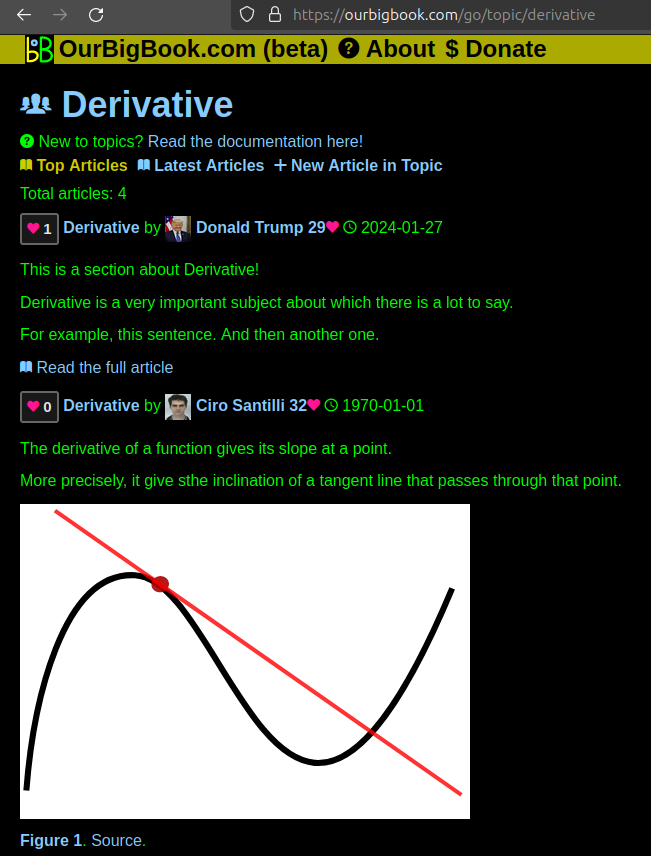
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.
Figure 4. Visual Studio Code extension tree navigation.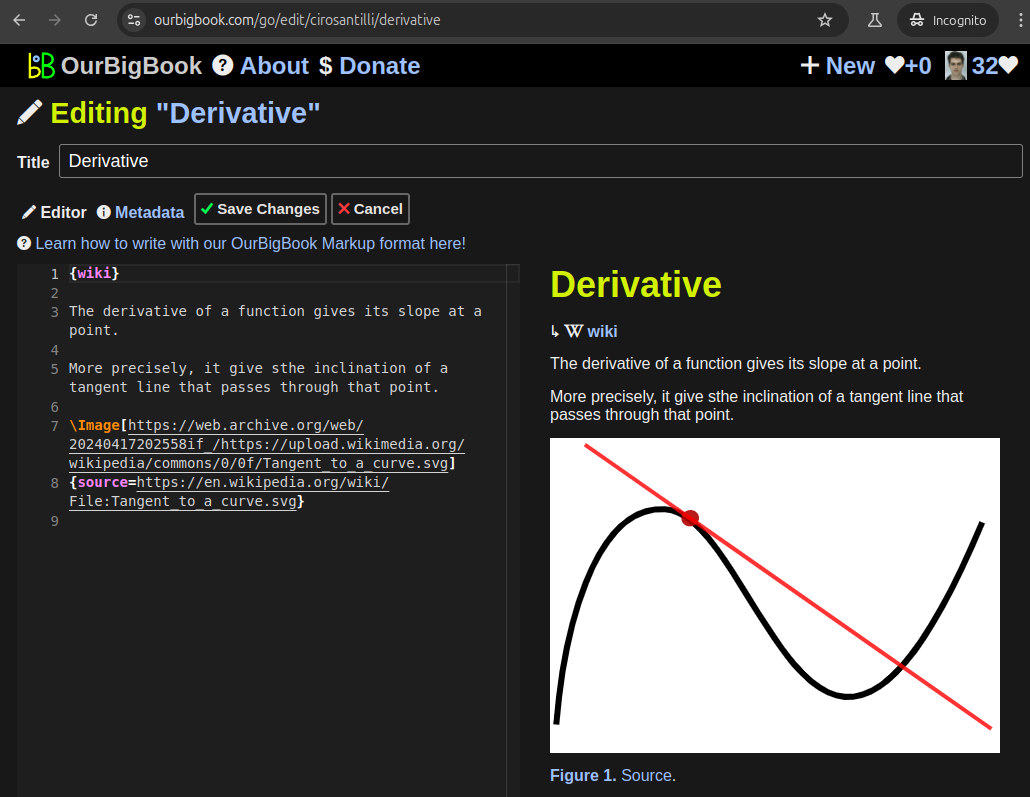
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 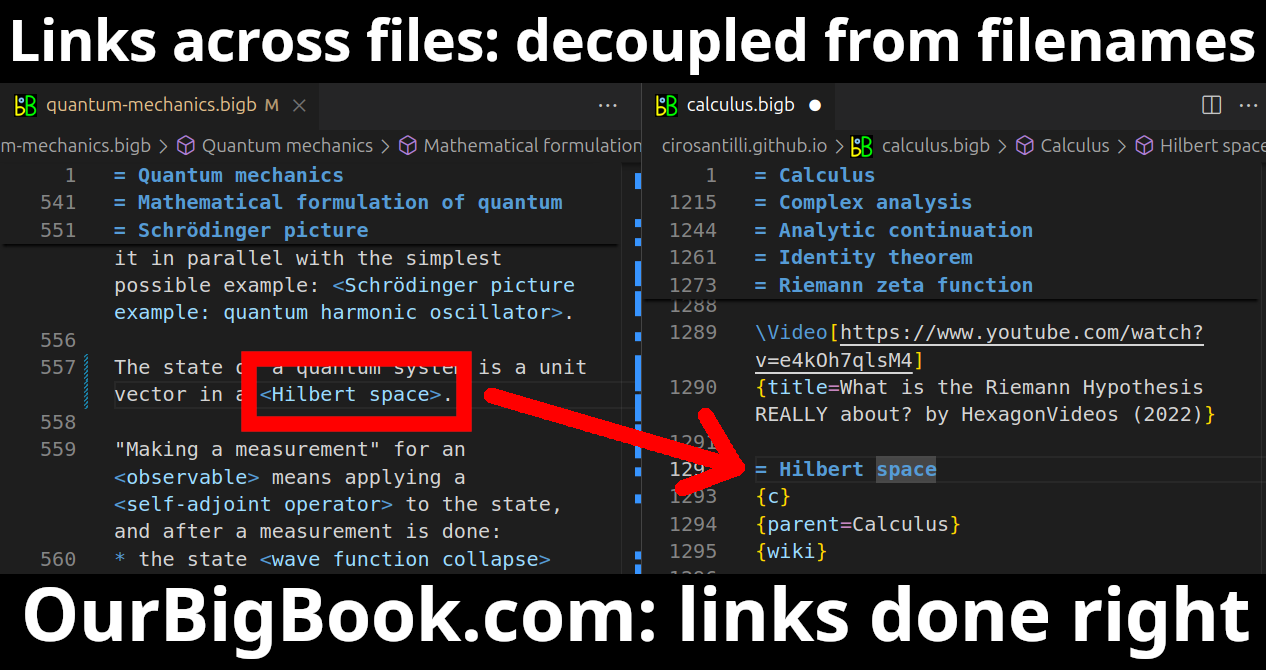
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact