"Holon" is a sculpture created by the artist Anthony Caro in 1968. This work is notable for its abstract form and use of steel, characteristic of Caro’s style, which often involved large, playful metal structures that interacted with their surroundings. The sculpture is an example of modernist art, moving away from representational forms and focusing on the interplay of shape, space, and color.
SplitsTree is a software tool used for the analysis and visualization of phylogenetic relationships and evolutionary processes. It is particularly known for its ability to construct and analyze various types of phylogenetic networks, which can represent complex evolutionary scenarios, including horizontal gene transfer, hybridization, and other non-tree-like evolutionary events.
Holmström's theorem, named after the economist Bengt Holmström, is a result in the field of contract theory. It revolves around the design of contracts in situations where there is asymmetric information, specifically regarding effort or actions taken by agents that cannot be perfectly observed by the principal. The key insights from Holmström's theorem are: 1. **Incentive Compatibility**: The theorem underscores the importance of designing contracts that provide the right incentives for agents (e.g.
A homothetic vector field is a type of vector field in differential geometry and the study of Riemannian manifolds that encodes self-similarity characteristics of the manifold. More precisely, a vector field \( V \) on a Riemannian manifold \( (M, g) \) is said to be homothetic if it generates homotheties of the metric \( g \).
The Hopf fibration is a mathematical construction that represents a particular way of decomposing certain spheres into circles. Named after Heinz Hopf, who introduced the concept in 1931, it provides a fascinating connection between topology, geometry, and algebra. Specifically, the Hopf fibration describes a fibration of the 3-sphere \( S^3 \) over the 2-sphere \( S^2 \) with the fibers being circles \( S^1 \).
Horizontal resistance refers to a level in a financial asset's price chart where the price tends to stop rising and may reverse direction. This is often seen as a price level where a significant number of sellers enter the market, causing the price to struggle to move above that level. In technical analysis, horizontal resistance is represented visually by a horizontal line drawn across the peaks in a price chart, indicating areas where the price has historically struggled to break through.
The Howland Will Forgery Trial refers to a legal case involving accusations of will forgery associated with the estate of an individual named Howland.
HTR3E refers to a specific type of serotonin receptor known as the 5-HT3 receptor, subtype E. Serotonin receptors are a class of receptors that bind serotonin (5-HT) and are involved in a variety of physiological and pharmacological processes. The 5-HT3 receptor is a ligand-gated ion channel that, when activated by serotonin, allows the flow of ions, which can lead to neuronal excitation.
Hubert Bray is a mathematician known for his work in the fields of differential geometry and general relativity, particularly in the study of geometric analysis. He has made significant contributions to topics such as minimal surfaces and the study of the topology of manifolds.
A humbucker is a type of electric guitar pickup designed to eliminate the electrical hum and noise that can be picked up by single-coil pickups. The name "humbucker" comes from its primary function: it "bucks" or cancels out the hum that is often introduced by electromagnetic interference. Humbuckers typically consist of two coils of wire wound around a magnetic core, positioned close to each other. These coils are wired in such a way that their outputs are out of phase.
Humiliation is the act of making someone feel ashamed, embarrassed, or degraded, often by putting them in a situation that lowers their dignity or self-esteem. It can occur in various contexts, such as social interactions, workplaces, academic settings, or intimate relationships. The experience of humiliation can be deeply impactful, leading to feelings of inferiority, worthlessness, or distress. It can result from intentional actions by others or can be a consequence of certain social dynamics or environments.
Hurricane Lorenzo was a powerful tropical cyclone that occurred in the Atlantic Ocean in September and October 2019. It was notable for being one of the strongest hurricanes to form in the eastern part of the Atlantic Ocean. Lorenzo originated from a tropical wave that moved off the coast of Africa and developed into a tropical storm on September 28, 2019.
A hybridization probe is a nucleic acid sequence, typically a DNA or RNA strand, that is used to detect the presence of complementary sequences in a sample. These probes are often used in molecular biology techniques such as PCR (polymerase chain reaction), Southern blotting, Northern blotting, and fluorescence in situ hybridization (FISH).
A hybrid market is a marketplace that combines elements of both traditional physical commerce and digital or online commerce. This structure allows businesses and consumers to interact and transact using multiple channels, such as in-person sales, e-commerce, mobile apps, and digital platforms. ### Key Characteristics of Hybrid Markets: 1. **Integration of Channels**: Hybrid markets leverage both online and offline channels, allowing consumers to choose their preferred method of purchasing.
HYDAC is a global company based in Germany that specializes in the development, manufacturing, and distribution of products and systems for fluid power, automation, and environmental technologies. Founded in 1963, HYDAC focuses on hydraulic and lubrication systems, as well as various components such as filters, accumulators, sensors, and cooling systems. The company's products are widely used in various industries, including construction machinery, mobile applications, industrial machinery, and energy technology.
A hygrometer is an instrument used to measure the humidity, or moisture content, in the air. It can provide readings in various units, such as relative humidity percentage, absolute humidity, or dew point temperature. There are several types of hygrometers, including: 1. **Mechanical Hygrometers**: These use materials that expand or contract with moisture levels, often represented by a dial or needle.
A **hyperperfect number** is a generalization of perfect numbers. While a perfect number is defined as a positive integer that is equal to the sum of its proper divisors (excluding itself), hyperperfect numbers extend this concept by introducing a parameter. In particular, a hyperperfect number can be defined in relation to a positive integer \( k \).
I/O scheduling refers to the method by which an operating system determines the order in which I/O operations are processed. It involves managing the access to input/output devices—such as hard drives, network interfaces, and other peripherals—to optimize system performance, resource utilization, and responsiveness. Key objectives of I/O scheduling include: 1. **Maximizing Throughput**: Ensuring the highest number of I/O operations are completed in a given time frame.
"Irish measure" refers to a system of measurement traditionally used in Ireland, often for various agricultural and trade purposes. It includes units for measuring quantities of grain, land, and other goods. Some examples of Irish measures include: 1. **Barley measure**: Used for measuring grain, where a barrel could hold a specific quantity of barley. 2. **Acres and roods**: Traditional units for measuring land area, with an acre being a standard unit.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.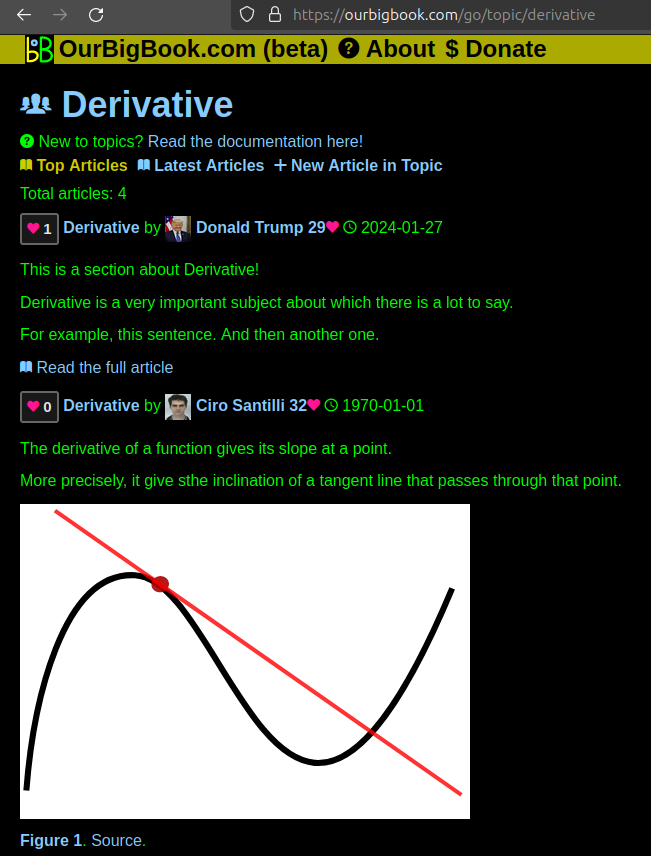
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.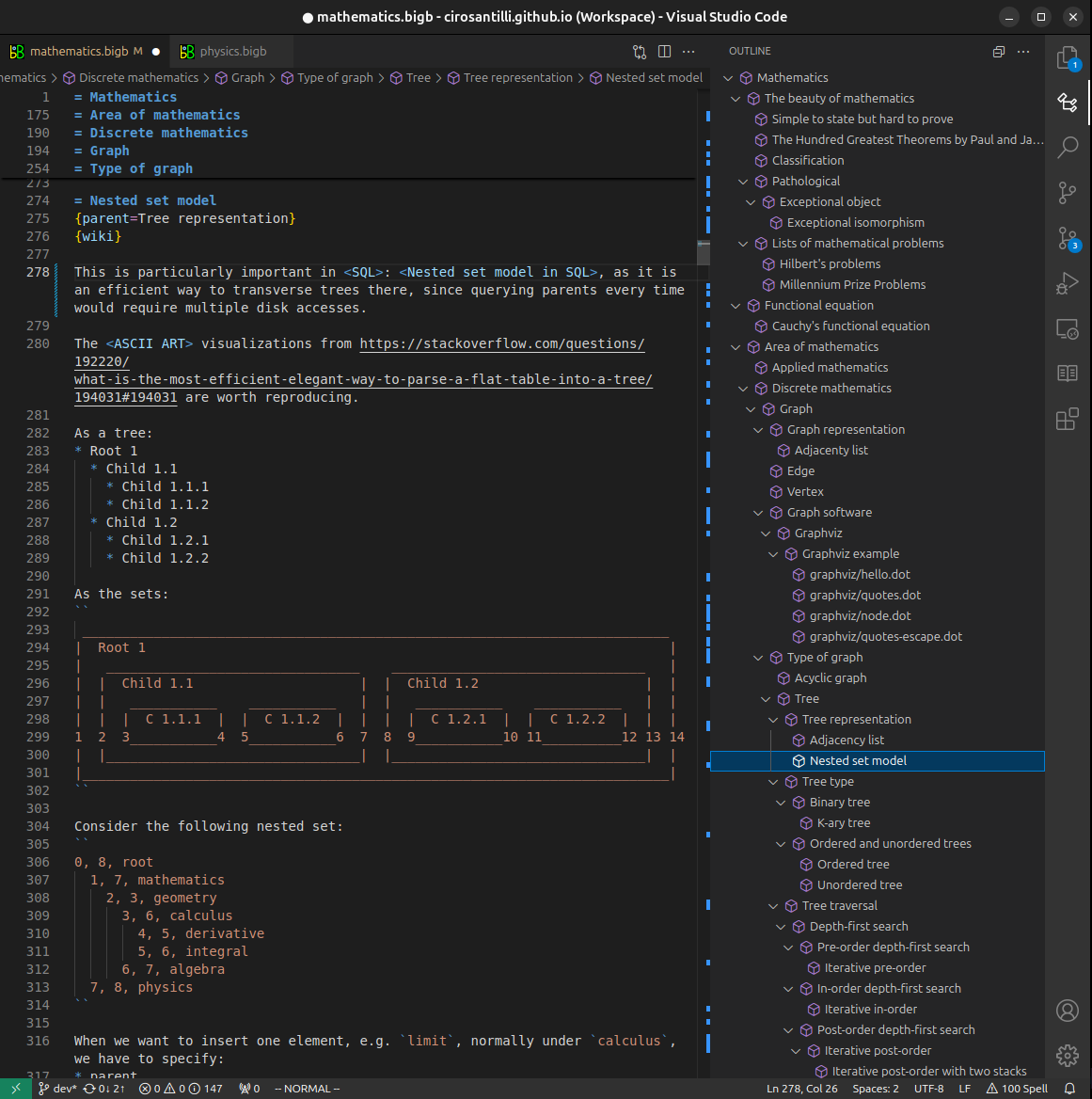
Figure 4. Visual Studio Code extension tree navigation.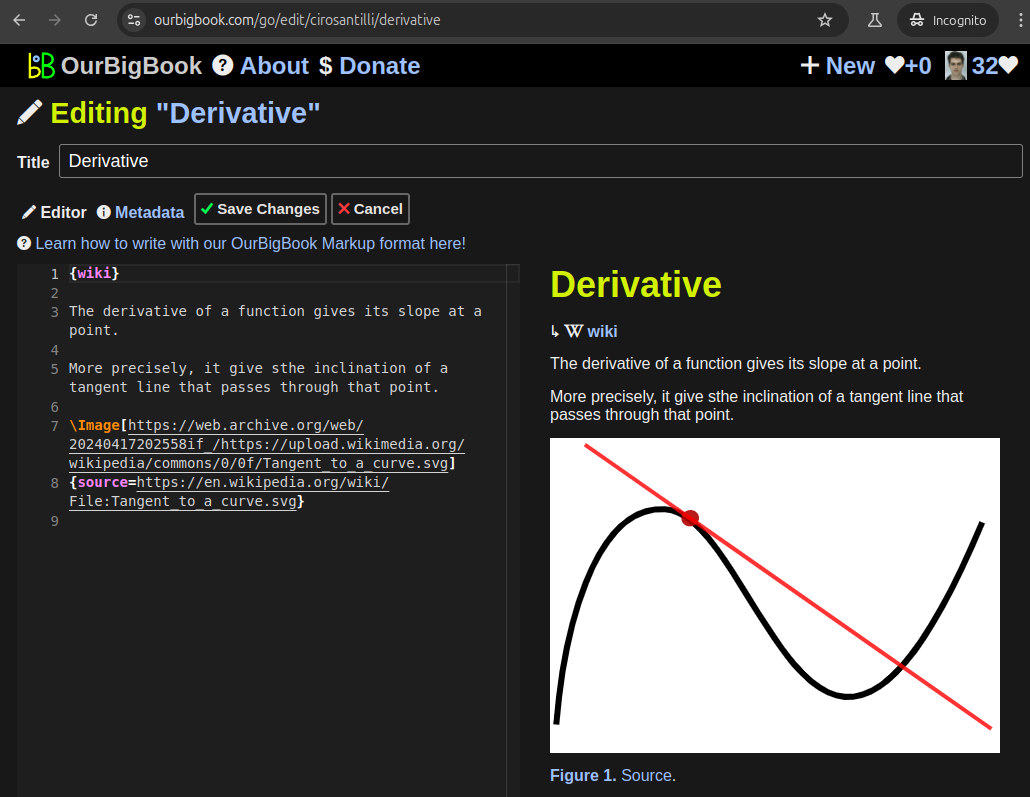
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 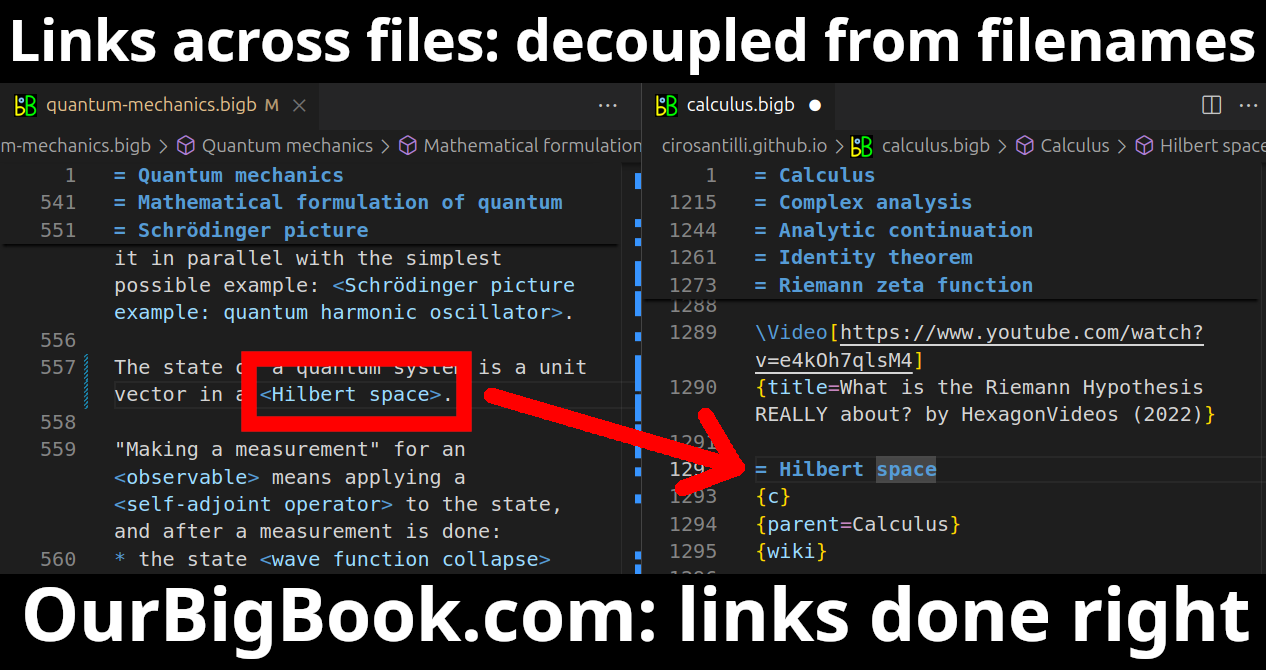
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact