Topological quantum numbers are integer values that arise in the context of topological phases of matter and quantum field theories, particularly in condensed matter physics. They characterize different phases of a system based on their global properties rather than local properties, which can be crucial for understanding phenomena that are stable against local perturbations. A few key points about topological quantum numbers are: 1. **Robustness**: Topological quantum numbers are robust against small perturbations or changes in the system.
Michael Trick is a well-known figure in the field of operations research and management sciences. He is a professor at Carnegie Mellon University, where he has contributed significantly to optimization, especially in the areas of integer programming and combinatorial optimization. His work often involves developing algorithms and computational methods to solve complex decision-making problems. In addition to his academic contributions, Trick is also recognized for his involvement in the operations research community, including organizing conferences and workshops.
Gustav Fechner (1801–1887) was a German philosopher, physicist, and psychologist who is best known for founding psychophysics, a field that explores the relationships between physical stimuli and the sensations and perceptions they produce. Fechner’s work laid the groundwork for modern experimental psychology. One of his most significant contributions was the formulation of Fechner's Law, which quantifies the relationship between the intensity of a stimulus and the resulting sensation.
Horatio Pollock is not a widely recognized name in mainstream culture or academia, and there may be no figures of notable prominence by that exact name as of my last knowledge update in October 2023. It’s possible that the name could refer to a lesser-known individual or be used in a specific context such as literature, a fictional character, or an obscure historical figure.
Klaus Kubinger is an Austrian psychologist and statistician known for his work in the fields of psychological measurement and test theory. He has contributed to various aspects of psychological assessment, including the development and validation of tests and measures used in psychological research and practice.
ISO 10303, commonly known as STEP (Standard for the Exchange of Product model data), is an international standard for the representation and exchange of product model data. It is widely used in various industries, especially in manufacturing, engineering, and product design, facilitating a wide range of applications such as computer-aided design (CAD), computer-aided manufacturing (CAM), and product lifecycle management (PLM).
The USC-Lockheed Martin Quantum Computing Center is a collaborative facility that aims to advance research and development in quantum computing technologies. Established through a partnership between the University of Southern California (USC) and Lockheed Martin, the center serves as a hub for academic researchers and industry professionals to work together on quantum computing projects and applications.
Encompassment ordering is a concept often discussed in the context of formal semantics, particularly within linguistics and logic. It relates to the way certain expressions can represent or capture a hierarchical relationship between sets or propositions. In general, an "encompassed" set is one that is contained within another set; therefore, an encompassing order reflects a hierarchy where certain elements or propositions are subordinate to others.
Longevity myths refer to common misconceptions and beliefs about aging and how to achieve a long life. These myths may stem from cultural norms, anecdotal evidence, or misinterpretations of scientific studies. Here are some examples of longevity myths: 1. **Myth: Genetics is the only factor in longevity.** - While genetics do play a role in how long we live, lifestyle factors like diet, exercise, social connections, and environmental influences are equally, if not more, important.
Internet case law refers to legal rulings and precedents that stem from disputes, conflicts, and issues specifically related to the use of the Internet and digital technologies. These cases can cover a wide range of topics, including: 1. **Intellectual Property**: Issues related to copyright, trademark, and patent infringement online, such as unauthorized sharing of digital content or the use of trademarks in domain names.
Internet governance organizations are entities that develop policies, standards, and guidelines aimed at managing and regulating various aspects of the Internet. These organizations are responsible for addressing technical, legal, economic, and social issues related to the Internet. Their roles can range from setting technical protocols that ensure interoperability to establishing frameworks for online conduct and data privacy.
A **summation generator** generally refers to a tool or software component that is designed to produce a summation (or series) of numerical values based on a defined mathematical expression or set of criteria. This can involve various scenarios and applications, ranging from basic arithmetic to more complex calculus operations. ### In Mathematics and Programming 1.

Composed mostly of the Virgo cluster and the Local group.
The "Most Vexing Parse" is a term used in C++ to describe an ambiguity that arises in the language when the compiler encounters certain declarations. It specifically refers to the situation where an expression can be interpreted in more than one way, leading to confusion about the intent of the programmer. One common example occurs when a function is declared with a certain syntax that can be interpreted either as a function declaration or as an object of a type being instantiated.
The medieval Islamic world, spanning roughly from the 8th to the 14th centuries, was a golden age for mathematics and science, marked by significant developments that were influenced by earlier Greek, Indian, and Persian knowledge but also included original contributions. Here are some key mathematicians and their contributions from this period: 1. **Al-Khwarizmi (c.
Maria Assumpció Català i Poch is a prominent figure known primarily in the fields of education and research in Catalonia, Spain. While specific details about her contributions can vary, she is associated with advancements in educational methodologies, possibly focusing on areas such as language education, curriculum development, or pedagogical strategies.
As of my last knowledge update in October 2021, "Lupitus of Barcelona" does not appear to be a widely recognized term or entity. It's possible that it could refer to a specific event, organization, product, or cultural reference that has emerged after that date. Alternatively, it might be an obscure term or a typographical error for something else.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.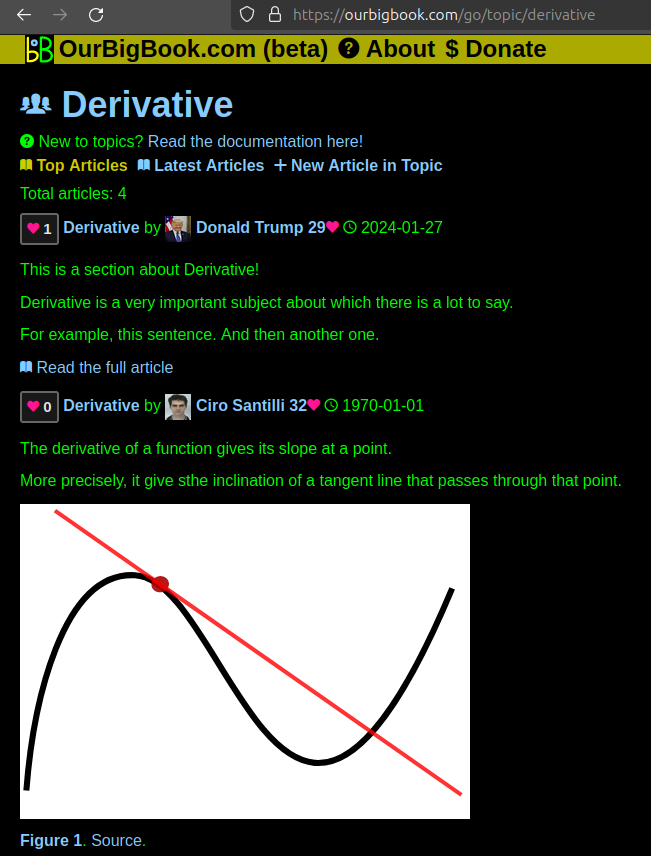
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.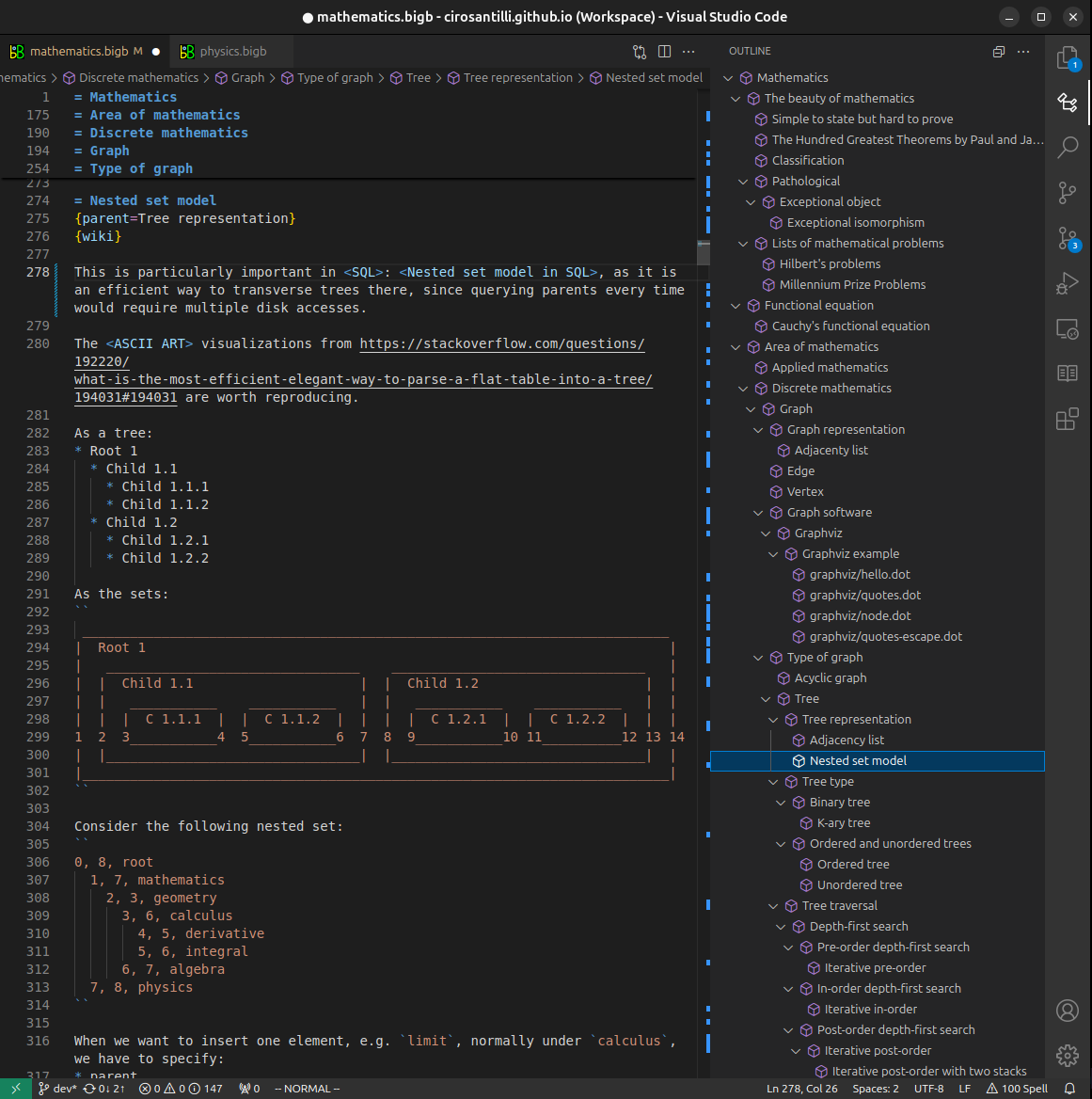
Figure 4. Visual Studio Code extension tree navigation.
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 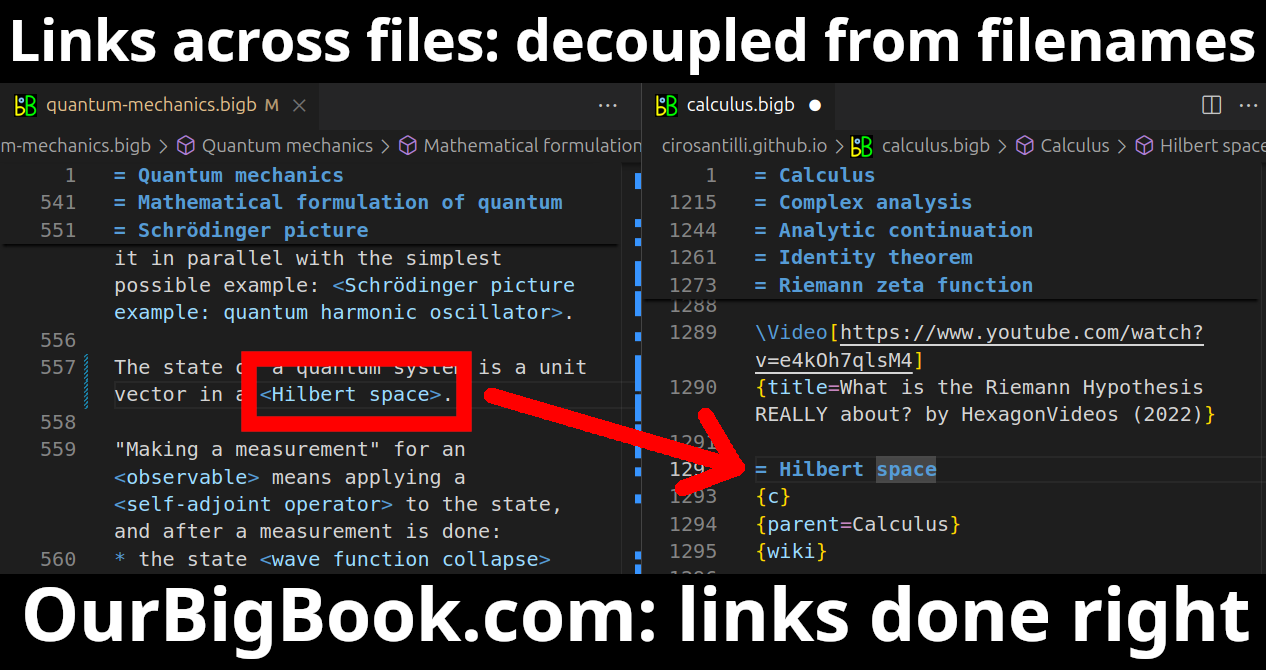
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact