In 1970, several computer companies were disestablished or went out of business as the technology landscape rapidly evolved. Some notable examples include: 1. **Data General Corporation** - Founded in 1968, Data General was known for its early minicomputers. While it was not completely disestablished until later, it faced significant challenges and declined in the early 1970s due to competition from larger companies like IBM.
Several computer companies were disestablished or ceased operations in 1971, although detailed records may be less comprehensive than for larger, more well-known companies. One of the more notable disestablishments in that year was **Bendix Corporation's computer division**, which was integrated into Allied Signal after a series of mergers and acquisitions.
In 1973, a number of computer companies were disestablished or ceased operations, though the specifics can vary based on the context and the regions involved. One notable example is: 1. **General Motors Research Laboratories (GM Research)**: While primarily an automotive company, GM was heavily involved in computing and technology development during the late 1960s and early 1970s.
Reza Olfati-Saber is a prominent researcher and academic known for his work in the fields of control theory, optimization, and networked systems, particularly in relation to distributed and multi-agent systems. He has made significant contributions to topics such as consensus algorithms, cooperative control, and networked robotics. His academic background includes a Ph.D. in Electrical Engineering, and he has held various positions in academia and research institutions.
In 1978, several computer companies were disestablished due to various reasons such as mergers, acquisitions, or financial difficulties. One notable example is: - **Computer Automation, Inc.** was a company that specialized in computer automation and industrial computing but was ultimately disbanded or restructured around this time. Keep in mind that details might vary, and the landscape of the computer industry was rapidly changing during that period, leading to the rise and fall of various companies.
"Annales Henri Poincaré" is a mathematical journal that publishes original research articles in various fields of mathematics and theoretical physics. Named after the renowned French mathematician Henri Poincaré, the journal aims to foster the dissemination of high-quality research in areas such as dynamical systems, mathematical physics, probability theory, and more. It is known for its rigorous peer-review process and its focus on the interplay between mathematics and its applications in physical sciences.
Electro-olfactography (EOG) is a technique used to study the olfactory system, or sense of smell, by measuring the electrical responses of the olfactory mucosa when exposed to odorants. This method involves placing electrodes on the olfactory epithelium (the tissue responsible for detecting odors) to record changes in electrical activity as the epithelium interacts with specific odor molecules.
Blood pressure is the force exerted by circulating blood against the walls of blood vessels, particularly the arteries. It is an essential measure of cardiovascular health and is expressed in terms of two readings: 1. **Systolic Pressure**: This is the higher number and represents the pressure in the arteries when the heart beats and pumps blood. 2. **Diastolic Pressure**: This is the lower number and indicates the pressure in the arteries when the heart is at rest between beats.
A "List of variations on a theme by another composer" typically refers to musical compositions that are variations on a theme originally written by a different composer. This form is popular in classical music, where composers take an existing melody and create a new work based on it, often developing it through various compositional techniques. Here are some notable examples: 1. **Variations on a Theme by Haydn, Op.
Blood volume refers to the total amount of blood in the circulatory system of a person or an animal. It is typically expressed in liters or milliliters and varies depending on factors such as body size, age, gender, and overall health. In an average adult, blood volume is approximately 5 to 6 liters. This accounts for about 7% to 8% of total body weight.
The Henderson–Hasselbalch equation is a fundamental equation in biochemistry and pharmacology that relates the pH of a solution to the pKa of an acid and the ratio of the concentration of its dissociated (conjugate base) and undissociated (acid) forms. It is often used to estimate the pH of buffer solutions.
"Inventiones Mathematicae" is a well-known mathematics journal that publishes research papers in all areas of mathematics. Established in 1966, it is a peer-reviewed journal and is recognized for its high standards of quality in mathematical research. The journal covers a wide range of topics, including but not limited to pure mathematics, applied mathematics, and mathematical theories. It aims to disseminate original work that contributes significantly to the field.
Survival analysis is a branch of statistics focused on analyzing the time until an event of interest occurs. This event is often referred to as a "failure" or "death," although it can represent any type of event, such as recovery from a disease, mechanical breakdown, or customer churn. Key concepts in survival analysis include: 1. **Survival Time**: The duration until the event occurs. This can be measured in various units, such as days, months, or years.
Philosophy of mathematics journals are academic periodicals that publish research on topics related to the philosophical foundations, implications, and interpretations of mathematics. These journals explore various issues, such as the nature of mathematical objects, the meaning of mathematical truth, the relationship between mathematics and the physical world, the processes of mathematical reasoning, and the epistemology and ontology of mathematical knowledge.
Probability journals are academic publications that focus on research in the field of probability theory and its applications. These journals typically publish original research articles, surveys, and reviews that contribute to the theoretical foundations of probability, as well as its applications in various fields such as statistics, finance, engineering, physics, and more. Some key features of probability journals include: 1. **Research Articles**: These journals publish peer-reviewed articles that present new findings, methodologies, or breakthroughs in probability theory.
Acta Mathematicae Applicatae Sinica is a scientific journal that focuses on applied mathematics. It publishes research articles, reviews, and other contributions related to various areas of applied mathematics, including computational mathematics, mathematical modeling, and numerical analysis. The journal aims to disseminate significant advancements and findings in applied mathematics, particularly those relevant to real-world problems and applications. It typically features work from researchers in the field, promoting the exchange of ideas and fostering collaboration among mathematicians and scientists.
"Algorithms" is a scholarly journal that focuses on the study and application of algorithms in various fields, including computer science, mathematics, and engineering. It publishes original research articles, reviews, and surveys that address both theoretical aspects and practical applications of algorithms. The journal covers a broad range of topics, such as algorithm design, complexity theory, data structures, computation models, and applied algorithms in areas like data mining, artificial intelligence, and optimization.
Axioms is an interdisciplinary mathematics journal that provides a platform for the publication of original research articles, surveys, and reviews covering a wide array of topics within the field of mathematics. The journal aims to promote high-quality research and foster communication among mathematicians working in different areas. Axioms is known for its open-access model, allowing researchers and the public to access published papers free of charge. This model helps disseminate mathematical knowledge more broadly and supports the idea of open science.
The Bulletin of Mathematical Sciences is an academic journal that publishes research articles, survey papers, and other mathematical content. Established to promote and disseminate important developments in the field of mathematics, the journal typically features articles covering a wide range of mathematical topics, including pure mathematics, applied mathematics, and interdisciplinary studies involving mathematics. The aim of the Bulletin is to provide a platform for mathematicians to share their findings, review current trends, and highlight significant advancements in the discipline.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.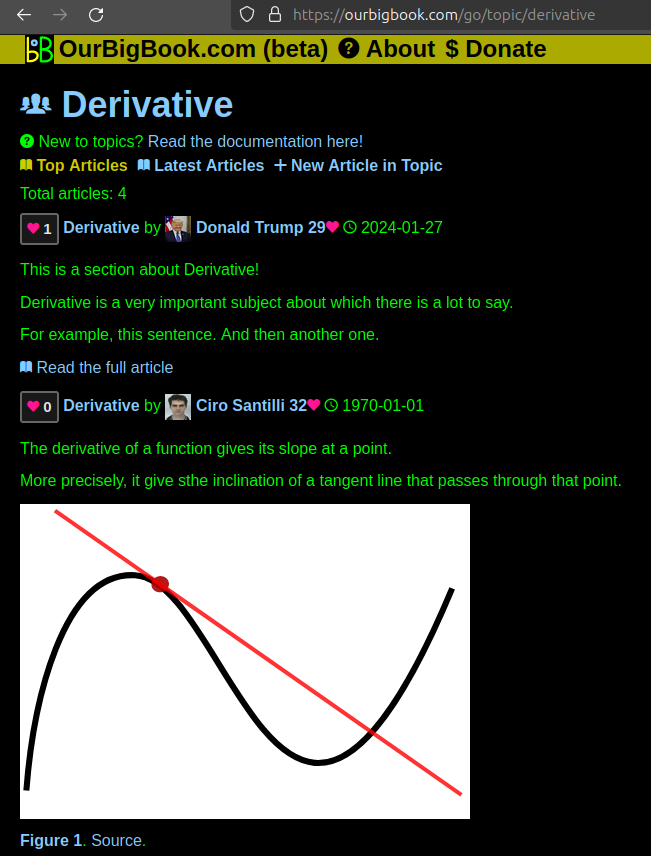
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.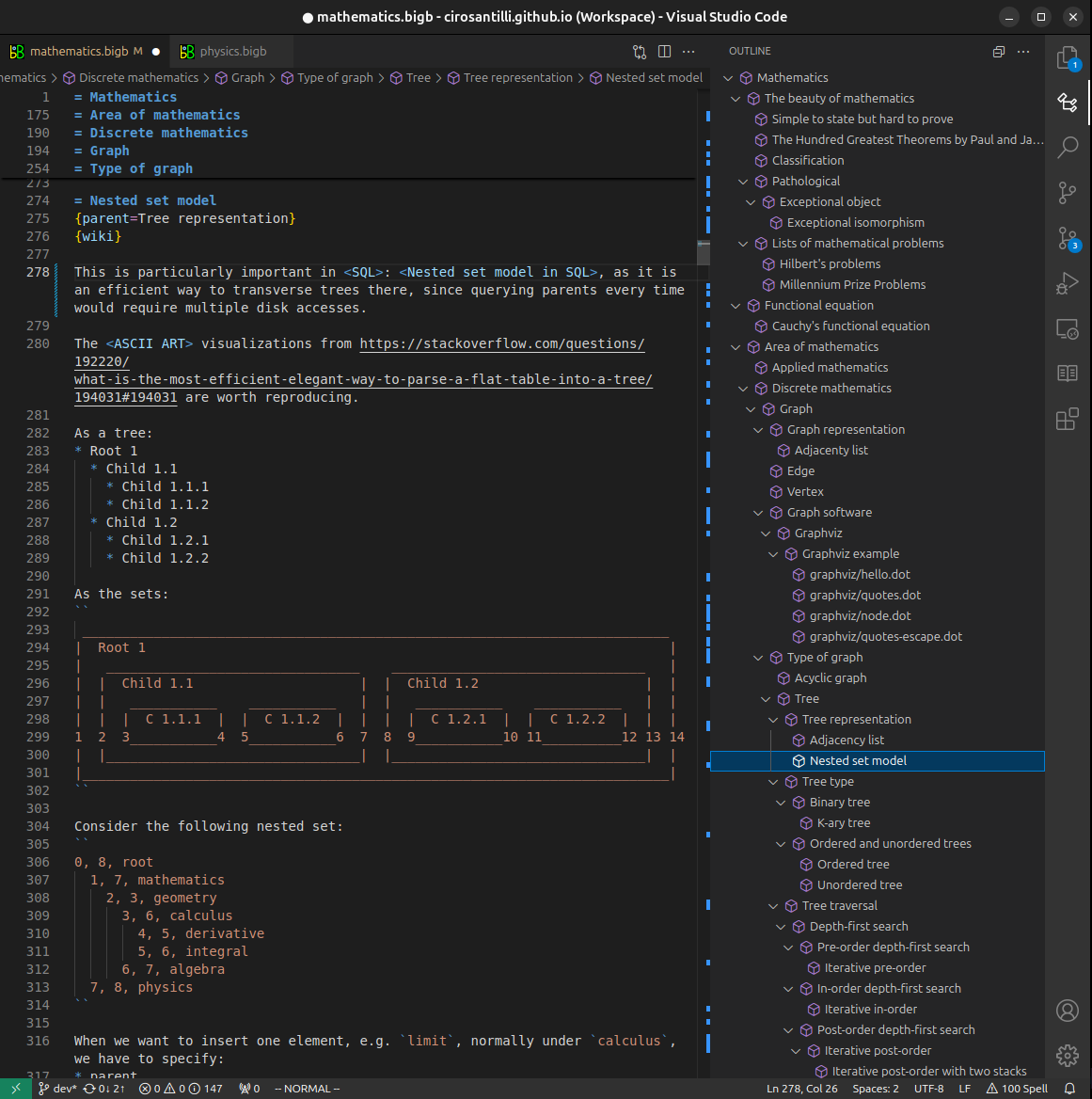
Figure 4. Visual Studio Code extension tree navigation.
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 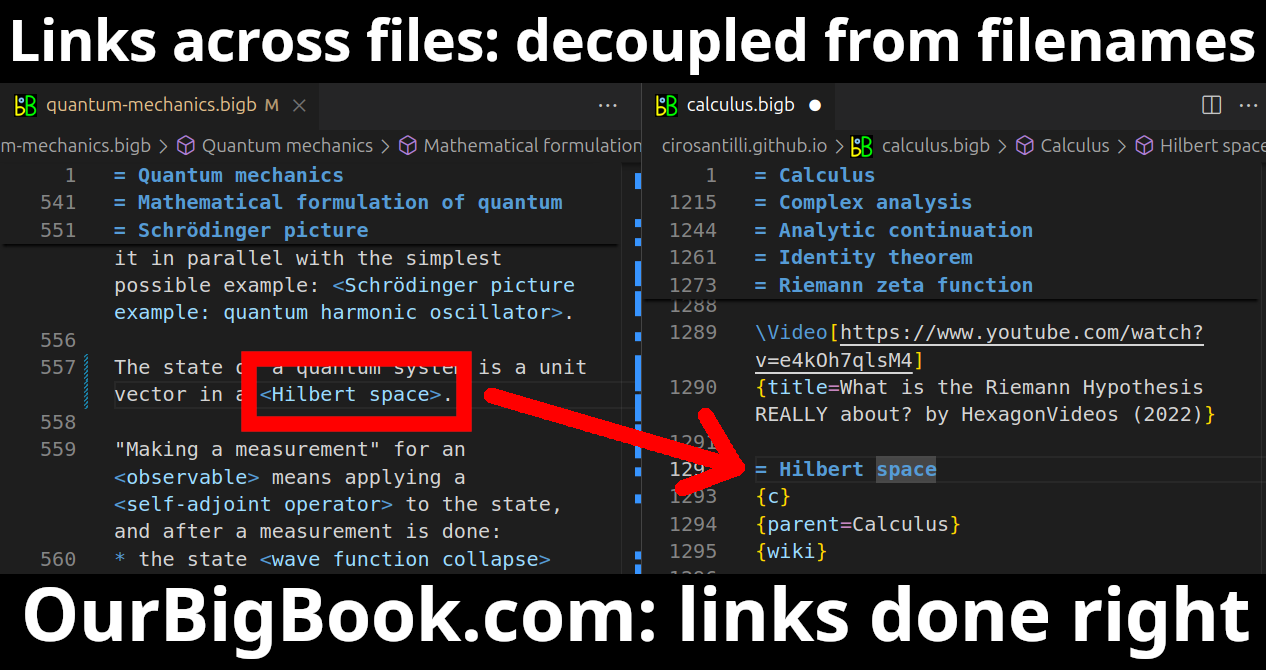
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact