The Glauber–Sudarshan P representation is an important tool in quantum optics and quantum mechanics for describing the statistical state of a quantum system, particularly in the context of light and bosonic fields. This representation provides a way to express the density operator (or state) of a quantum system as a distribution over the phase space of classical probabilities. ### Key Concepts 1.
The Mandel Q parameter is a measure used in quantum optics to quantify the non-classicality of light. It is defined in terms of the number of photons in a given mode of light and refers to the degree of deviation of photon number statistics from that expected for classical light sources.
Fractions in mathematics represent a way to express a part of a whole. A fraction consists of two main components: 1. **Numerator**: The number on the top, which indicates how many parts you have. 2. **Denominator**: The number on the bottom, which indicates how many equal parts the whole is divided into.
Graphene is a single layer of carbon atoms arranged in a two-dimensional honeycomb lattice. It is known for its remarkable electrical, thermal, and mechanical properties. Here are some key characteristics and applications of graphene: ### Properties: 1. **Strength**: Graphene is extremely strong—about 200 times stronger than steel—yet very lightweight. 2. **Electrical Conductivity**: It has exceptional electrical conductivity, making it conducive for electronic applications.
A strained quantum-well laser (SQWL) is a type of semiconductor laser that utilizes quantum wells under strain to enhance performance characteristics. Quantum wells are thin layers of semiconductor material where charge carriers (electrons and holes) are confined in one dimension, leading to quantized energy levels. In a strained quantum-well laser, the quantum wells are created within a lattice structure that is intentionally misaligned or geometrically altered.
Attila Grandpierre is a Hungarian physicist and philosopher known for his work in theoretical physics, particularly in areas related to the foundations of quantum mechanics and cosmology. He has also contributed to discussions on the intersection of science and philosophy. Grandpierre is recognized for his holistic approach to science, emphasizing the interconnectedness of various scientific disciplines and the philosophical implications of scientific theories.
Howard M. Wiseman is an Australian theoretical physicist known for his work in the fields of quantum physics, particularly quantum information and quantum measurement theory. He has contributed to various aspects of quantum mechanics, including research on quantum entanglement and the foundations of quantum theory. Wiseman has also been involved in developing the framework for quantum measurement and has made significant contributions to the understanding of the role of measurement in quantum systems. His work often encompasses both theoretical developments and potential applications in quantum technologies.
CloudMask is a specific cloud service or tool designed to assist in cloud-based data management, security, or processing tasks, particularly in the context of cloud storage or data analysis. It allows users to protect sensitive data by masking or anonymizing it, ensuring privacy and compliance with various regulations.
Ira N. Levine is a prominent American physicist known for his contributions to the field of physical chemistry and chemistry education. He is particularly recognized for his work on the principles of physical chemistry, which has been widely disseminated through his textbook, "Physical Chemistry." This textbook is often used in university courses and is appreciated for its clarity and comprehensive coverage of the subject. Levine has had a significant impact on both the academic community and students studying chemistry.
A dosimeter is a device used to measure an individual's exposure to ionizing radiation, such as gamma rays, X-rays, and beta particles. It is commonly used in environments where radiation exposure is a concern, such as in nuclear power plants, medical facilities, research laboratories, and during certain industrial processes. Dosimeters come in various forms, including: 1. **Film Badges**: These contain a photographic film that darkens in response to radiation exposure.
A battery eliminator circuit is an electronic circuit designed to provide the equivalent voltage and current of a battery to a device without actually using a physical battery. These circuits are often used in applications where a device typically powered by batteries can be operated continuously or when it is used in a fixed location, such as during testing, prototype development, or in applications where the cost or inconvenience associated with replacing batteries frequently is a concern.
Michelle Simmons is an Australian physicist known for her work in quantum computing and nanotechnology. She has made significant contributions to the development of silicon-based quantum computers and has been a prominent advocate for STEM (science, technology, engineering, and mathematics) education and increasing diversity in these fields. Simmons was awarded numerous honors throughout her career, including being named a fellow of the Australian Academy of Science and receiving the prestigious 2018 Australian of the Year award.
Peter R. Holland is a notable figure primarily recognized for his contributions to the field of zoology, particularly in the study of reptiles and amphibians. He is known for his work on the systematics and biogeography of various species, as well as his research on their evolutionary relationships.
Ross H. McKenzie is a prominent physicist known for his contributions to the fields of condensed matter physics and materials science. He has made significant advancements in understanding quantum mechanics and the behavior of materials at the atomic level. McKenzie has authored numerous research papers and has been involved in various academic and research activities. His work often explores the interactions between electron behavior and the properties of materials, contributing to advancements in both theoretical and experimental physics.
Samuel L. Braunstein is a prominent physicist known for his contributions to quantum mechanics and quantum information theory. He has made significant advancements in areas such as quantum computation, quantum communication, and quantum optics. His work often focuses on the theoretical foundations of these fields and the development of innovative technologies based on quantum principles.
Radio-controlled (RC) cars are miniature vehicles that are controlled remotely using a transmitter and receiver system. The transmitter is typically a handheld device equipped with controls allowing the operator to steer, accelerate, and brake the vehicle. The receiver is installed in the car and responds to the signals sent from the transmitter, controlling the car's movement.
Radioactive quackery refers to the promotion and use of products or treatments that claim to harness the benefits of radioactivity or radioactive materials without scientific backing or safety considerations. Historically, various individuals and companies have marketed radioactive substances, suggesting that they can cure illnesses or improve health, often exploiting the public's fascination and fear around radiation. This quackery can include items like radioactive water, radium-infused tonics, and other dubious health products.
A beta particle is a high-energy, high-speed electron or positron that is emitted during the radioactive decay of an atomic nucleus, a process known as beta decay. There are two types of beta particles: 1. **Beta-minus (β⁻) particles**: These are electrons emitted from a nucleus when a neutron is transformed into a proton. This process involves the release of an electron and an antineutrino.
A radio-controlled submarine is a model submarine that is operated remotely using a radio control system. These submarines can dive, surface, and maneuver underwater, making them an exciting option for hobbyists and enthusiasts interested in model boating. Key features of radio-controlled submarines include: 1. **Control System**: Typically, they use a transmitter (held by the operator) and a receiver (mounted in the submarine) to facilitate communication.
A remote control animal is a toy or device that mimics the movements and behaviors of real animals and can be controlled from a distance using a remote control. These devices are often designed for entertainment, education, or even companionship. Remote control animals can vary widely in terms of appearance, features, and functionalities. Some common types of remote control animals include: 1. **RC Dogs and Cats**: These toys can perform tricks, walk, bark, or meow, depending on their design.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.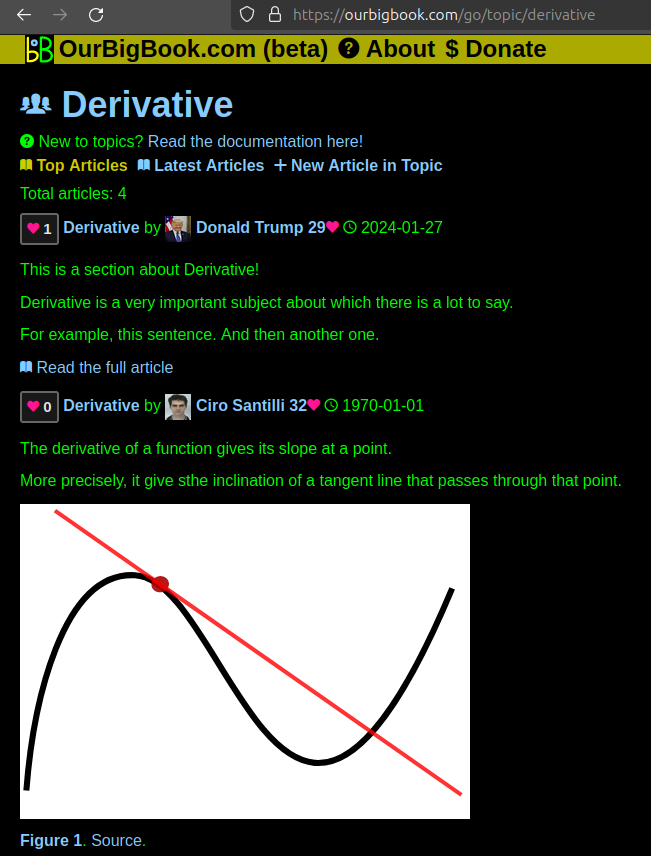
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.
Figure 4. Visual Studio Code extension tree navigation.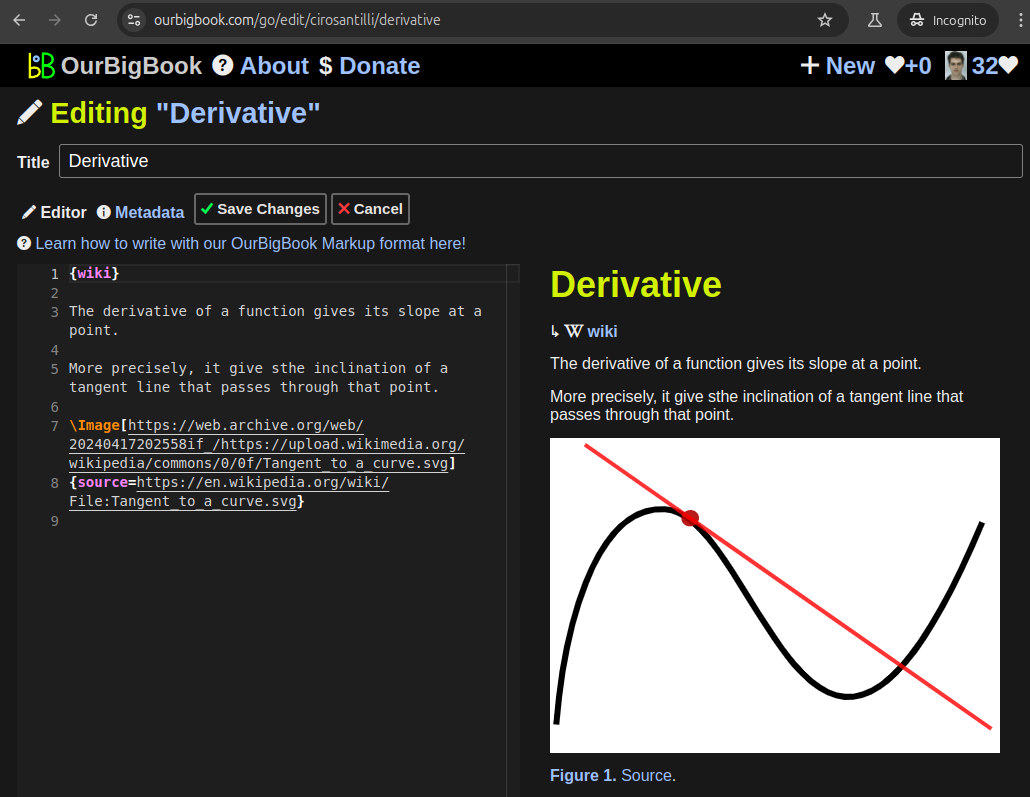
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 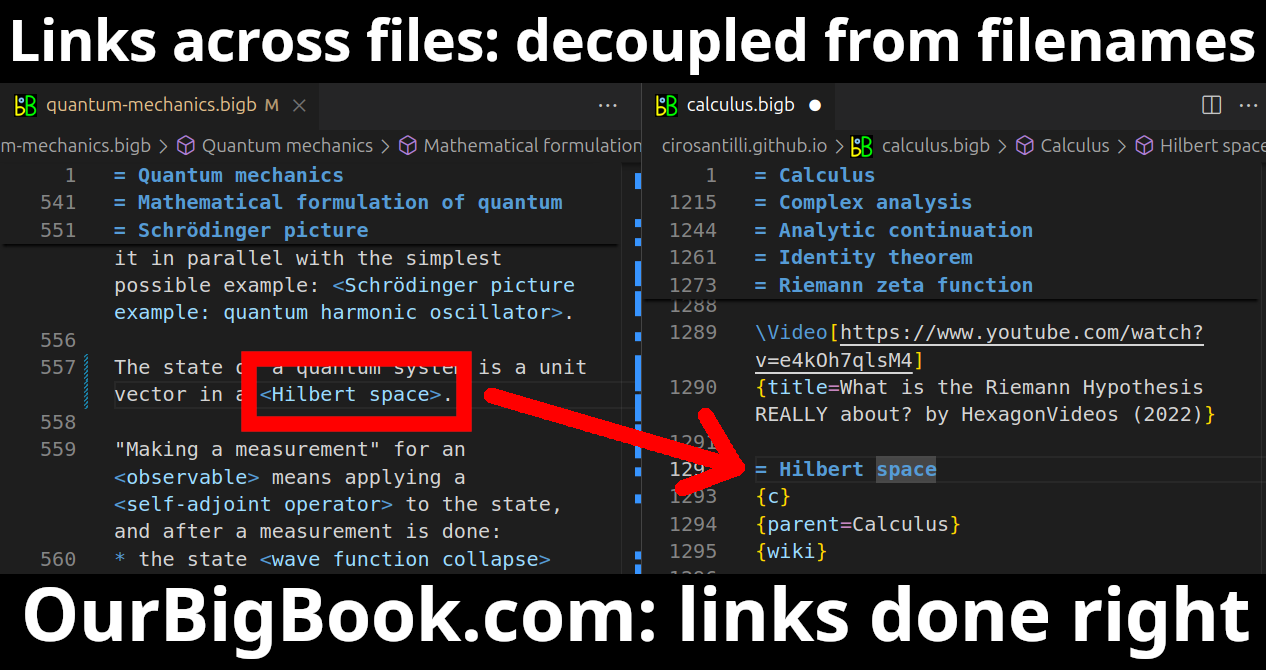
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact