Biorepositories, also known as biobanks, are facilities or collections that store biological samples, such as human tissue, blood, DNA, and other bodily fluids, as well as associated data. These samples are collected and stored for future research purposes, particularly in the fields of medicine, genetics, and biotechnology. Key aspects of biorepositories include: 1. **Sample Collection and Storage**: Biorepositories collect samples from donors, which may include healthy individuals or patients with specific conditions.
3D-Jury is a software application designed to facilitate the assessment and evaluation of projects in a three-dimensional space. It is often used in fields such as architecture, urban planning, and design to allow multiple stakeholders to review and provide feedback on 3D models or visualizations of projects. The platform enables users to interact with and manipulate 3D representations of projects collaboratively, which can enhance communication and decision-making during the project development process.
Martin Farach-Colton is a prominent computer scientist known for his contributions to algorithms, data structures, and bioinformatics. He has worked on various topics, including suffix trees, string algorithms, and the application of computational techniques to biological problems. Farach-Colton is also recognized for his role in academia, having served as a professor at institutions like Rutgers University. His work has significantly impacted theoretical computer science and has applications in areas such as genomics and data processing.
Algae DNA barcoding is a molecular technology used to identify and classify algal species based on short, standardized sequences of genetic material, typically from specific regions of their DNA.
Biclustering, also known as co-clustering or simultaneous clustering, is a data analysis technique that seeks to uncover patterns in data sets where both rows and columns are clustered simultaneously. Unlike traditional clustering methods, which typically group either rows (observations) or columns (features) independently, biclustering allows for the identification of subsets of data that exhibit similar characteristics across both dimensions.
BioPAX (Biological Pathway Exchange) is a standard format designed for the exchange, sharing, and representation of biological pathway information. It aims to enable interoperability among software and databases that manage biological data related to molecular interactions, cellular processes, and metabolic pathways. BioPAX provides a standardized vocabulary and structure for depicting biological entities—such as genes, proteins, and small molecules—and their interactions or relationships within biological pathways.
Biological network inference is the process of deducing or reconstructing biological networks from experimental data. These networks can represent various biological interactions and relationships, such as gene regulatory networks, protein-protein interaction networks, metabolic networks, and others. The goal of network inference is to understand the complex interactions that govern biological processes by creating models that illustrate how different components (genes, proteins, metabolites, etc.) interact with each other.
The Conference on Semantics in Healthcare and Life Sciences (CSHALS) is an academic and professional event that focuses on the application of semantic technologies in the fields of healthcare and life sciences. The conference typically brings together researchers, practitioners, and industry stakeholders to discuss the latest developments, research findings, and innovations related to semantic web technologies, knowledge representation, data interoperability, and data analytics within these domains.
The Darwin Core Archive (DwC Archive) is a data standard used for sharing biodiversity data. It is part of the Darwin Core standards, which provide a framework for providing information about biological diversity in a structured and interoperable way. The Darwin Core Archive facilitates the sharing and publishing of biodiversity datasets, particularly in the context of specimen records, observations, or related data concerning organisms. It consists of various types of metadata and data files that collectively allow for the easy exchange and usage of biodiversity information.
De novo transcriptome assembly is the process of reconstructing the complete set of RNA transcripts in a given organism or sample without prior reference to a known genome. This is particularly useful in situations where the genome of the organism is not available, poorly annotated, or when studying non-model organisms. Here are the key steps and concepts involved in de novo transcriptome assembly: 1. **RNA Extraction**: First, RNA is extracted from the cells or tissues of interest.
In bioinformatics, a dot plot is a graphical method used to visualize the similarities and differences between two biological sequences, such as DNA, RNA, or protein sequences. The primary purpose of a dot plot is to identify regions of similarity that may indicate homology, structural or functional relationships, or conserved sequences. ### How Dot Plots Work: 1. **Matrix Representation**: In a dot plot, one sequence is represented along the x-axis and the other along the y-axis.
Engineering biology is an interdisciplinary field that combines principles of biology, engineering, and computational sciences to design and manipulate biological systems for various applications. It encompasses a broad range of activities, including the development of synthetic biological systems, the design of new organisms, and the manipulation of existing biological functions for practical uses. Key aspects of engineering biology include: 1. **Synthetic Biology**: This involves designing and constructing new biological parts, devices, and systems, as well as redesigning existing biological systems for useful purposes.
ExPASy, or the Expert Protein Analysis System, is a bioinformatics resource portal operated by the Swiss Institute of Bioinformatics (SIB). It provides access to a variety of databases and tools for protein sequence analysis and functional annotation.
Fast statistical alignment is a computational method used in bioinformatics for aligning biological sequences, such as DNA, RNA, or protein sequences, quickly and efficiently. This technique is particularly useful when dealing with large datasets or when rapid results are needed for applications like phylogenetic analysis, comparative genomics, or sequence searching.
A Nexus file typically refers to a data file format used in various scientific fields, particularly in the context of imaging and data management. The term "Nexus" can be specifically associated with different disciplines, so its meaning may vary depending on the context.
Flux balance analysis (FBA) is a mathematical approach used in systems biology to analyze the flow of metabolites through metabolic networks. It is particularly useful for studying the metabolic pathways of microorganisms and for understanding how cells allocate resources between various biochemical processes. ### Key Features of Flux Balance Analysis: 1. **Metabolic Network Representation**: - Metabolic networks are typically represented as stoichiometric matrices, where the rows correspond to metabolites and the columns correspond to reactions.
GFP-cDNA refers to a complementary DNA (cDNA) that encodes the green fluorescent protein (GFP). GFP is a bioluminescent protein originally found in the jellyfish *Aequorea victoria*, and it emits a bright green fluorescence when exposed to ultraviolet or blue light. In molecular biology, cDNA is synthesized from messenger RNA (mRNA) through a process called reverse transcription.
Gene Set Enrichment Analysis (GSEA) is a computational method used to identify whether a predefined set of genes shows statistically significant differences in expression under different biological conditions, such as diseased versus healthy states or various treatments. The goal of GSEA is to determine whether genes that share common biological functions, chromosomal locations, or regulation are overrepresented (enriched) within a specific group of genes that have been identified as being significantly different between conditions.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.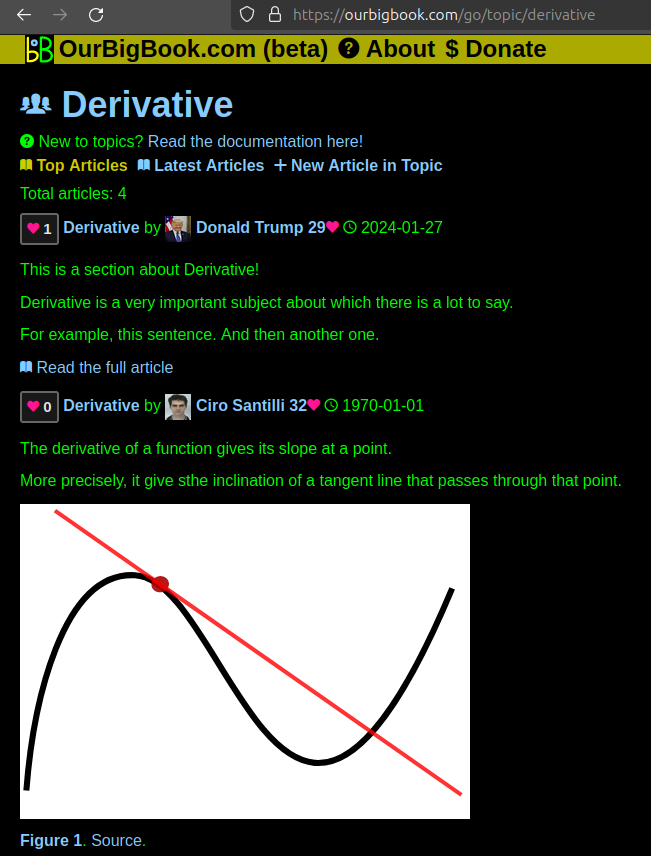
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control

Figure 2. You can publish local OurBigBook lightweight markup files to either OurBigBook.com or as a static website.
Figure 3. Visual Studio Code extension installation.
Figure 5. . You can also edit articles on the Web editor without installing anything locally. Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension. 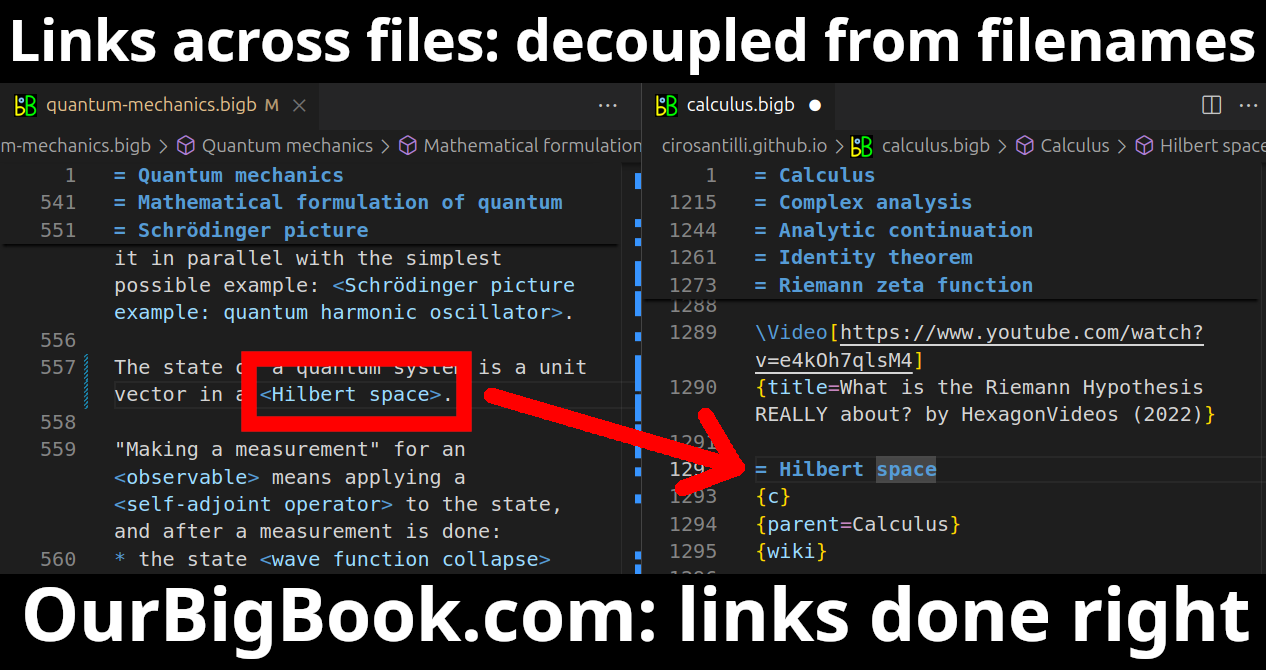
- Infinitely deep tables of contents:

All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact