Sequence transformation refers to various techniques or processes used to alter a sequence of elements, which can be numbers, characters, or other data types, in specific ways to achieve desired outcomes. This concept is commonly applied in several fields, including mathematics, computer science, data processing, and machine learning.
Variational perturbation theory is a method used in quantum mechanics and statistical mechanics to approximate the properties of a quantum system, particularly when dealing with a Hamiltonian that can be separated into a solvable part and a perturbation. The approach combines elements of perturbation theory with ideas from the variational principle, which is a powerful tool in quantum mechanics for approximating the ground state energy and wave functions of complex systems. ### Key Concepts 1.
Ewald summation is a mathematical technique used to compute the potential energy and forces in systems with periodic boundary conditions, commonly encountered in simulations of charged systems or dipolar systems in condensed matter physics, materials science, and molecular dynamics. The main challenge in these systems is that the Coulomb potential between charges, which falls off as \(1/r\), leads to divergent sums when calculated directly for an infinite periodic lattice.
A **subharmonic function** is a real-valued function that satisfies specific mathematical properties, particularly within the context of harmonic analysis and the theory of partial differential equations.
Scattering theory is a framework in quantum mechanics and mathematical physics that describes how particles or waves interact with each other and with potential fields. It is particularly important for understanding phenomena such as the collision of particles, where incoming particles interact with a potential and then emerge as outgoing particles. **Key Elements of Scattering Theory:** 1. **Scattering Process**: Involves an incoming particle (or wave) interacting with a target, which may be another particle or an external potential field.
BCFW recursion, or the Britto-Cachazo-Feng-Witten recursion, is a powerful technique in quantum field theory, particularly in the context of calculating scattering amplitudes in gauge theories and gravity. It was introduced by Fabio Britto, Freddy Cachazo, Bo Feng, and Edward Witten in the mid-2000s.
The Bunch-Davies vacuum is a concept in the context of quantum field theory, particularly in relation to the study of inflation in cosmology. It represents a specific vacuum state defined for quantum fields in de Sitter spacetime, which is the solution to Einstein's equations for a universe experiencing exponential expansion.
In physics, "crossing" typically refers to a specific phenomenon in the context of quantum mechanics or scattering theory. It is most commonly associated with the concept of **crossing symmetry**, which describes the relationship between different scattering processes. When particles collide, they can interact and scatter in various ways. The "crossing" concept allows physicists to relate different scattering processes to each other through transformations.
Dimensional reduction is a process used in data analysis and machine learning to reduce the number of random variables or features in a dataset while preserving its essential information. This is particularly useful when dealing with high-dimensional data, which can be challenging to visualize, analyze, and model due to the "curse of dimensionality" — a phenomenon where the feature space becomes increasingly sparse and less manageable as the number of dimensions increases.
A "dressed particle" is a concept used in quantum field theory and condensed matter physics. It refers to a particle that is "dressed" by its interactions with the surrounding environment, such as other particles, fields, or excitations. This idea contrasts with a "bare particle," which is an idealized version that doesn't account for such interactions.
False vacuum decay is a theoretical concept in quantum field theory and cosmology that describes a scenario in which a system exists in a metastable state (false vacuum) that is not the lowest energy state (true vacuum). In this context, the "false vacuum" is a local minimum of energy, but there exists a lower energy state, the "true vacuum," that the system can potentially transition into.
The Fermi point refers to a specific concept related to the behavior of quasi-particles in certain condensed matter systems, particularly in the context of topological materials and the study of fermionic systems. To understand the Fermi point, we can relate it to a few important concepts in solid-state physics and quantum field theory. 1. **Fermi Energy**: In solid-state physics, the Fermi energy is the highest energy level that electrons occupy at absolute zero temperature in a solid.
Four-fermion interactions refer to a type of interaction in quantum field theory where four fermions—particles that follow Fermi-Dirac statistics—interact with one another. Fermions include particles such as electrons, quarks, neutrinos, and their antiparticles. In a four-fermion interaction, two pairs of fermions interact simultaneously.
Intrinsic parity is a concept in particle physics that refers to a property of particles that characterizes their behavior under spatial inversion (or parity transformation). Parity transformation involves flipping the spatial coordinates, essentially transforming a point in space \((x, y, z)\) to \((-x, -y, -z)\). In terms of intrinsic parity, particles can be classified as having either positive or negative parity. This classification helps in understanding the symmetries and conservation laws of physical processes involving particles.
Quantum theory, also known as quantum mechanics, involves a variety of mathematical concepts and structures. Here’s a list of key mathematical topics that are often encountered in the study of quantum mechanics: 1. **Linear Algebra**: - Vector spaces - Inner product spaces - Operators (linear operators on Hilbert spaces) - Eigenvalues and eigenvectors - Matrix representations of operators - Schur decomposition and Jordan forms 2.
Magnetic catalysis refers to the process where magnetic fields enhance the rates of chemical reactions or facilitate certain transformations in materials. While the term can be associated with various contexts, it is especially relevant in fields like catalysis in chemistry and materials science. In the context of catalysis, magnetic materials or magnetic fields can influence the reactivity of catalysts or the kinetics of reactions.
Matsubara frequency is a concept commonly used in condensed matter physics and statistical mechanics, specifically in the context of finite-temperature field theory and many-body quantum systems. It arises in the formalism known as Matsubara techniques, which are used to evaluate correlations and Green's functions in systems at finite temperature. Matsubara frequencies are defined as discrete frequencies that appear in the solution of the equations describing quantum systems at finite temperature.
The Octacube is a large-scale sculpture created by artist Charles O. Perry. Composed of an intricate arrangement of interlocking forms, the piece is designed to evoke a sense of movement and energy. The sculpture often takes the shape of a cube, but its intricate structure and the way it is assembled can create a dynamic visual experience, where the viewer perceives different perspectives and angles as they move around it.
The Nielsen-Olesen string is a solution in theoretical physics that describes a type of magnetic string or vortex line that arises in certain gauge theories, particularly in the context of superconductivity and grand unified theories. It is named after Hans Christian Nielsen and Pierre Olesen, who first proposed these solutions in the early 1970s.
Non-invertible symmetry refers to a type of symmetry in physical systems where certain transformations cannot be undone or reversed. In contrast to invertible symmetries, which have a clear operation that can be applied to return a system to its original state, non-invertible symmetries do not allow for such a straightforward correspondence. This concept often arises in the context of condensed matter physics and quantum field theory.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.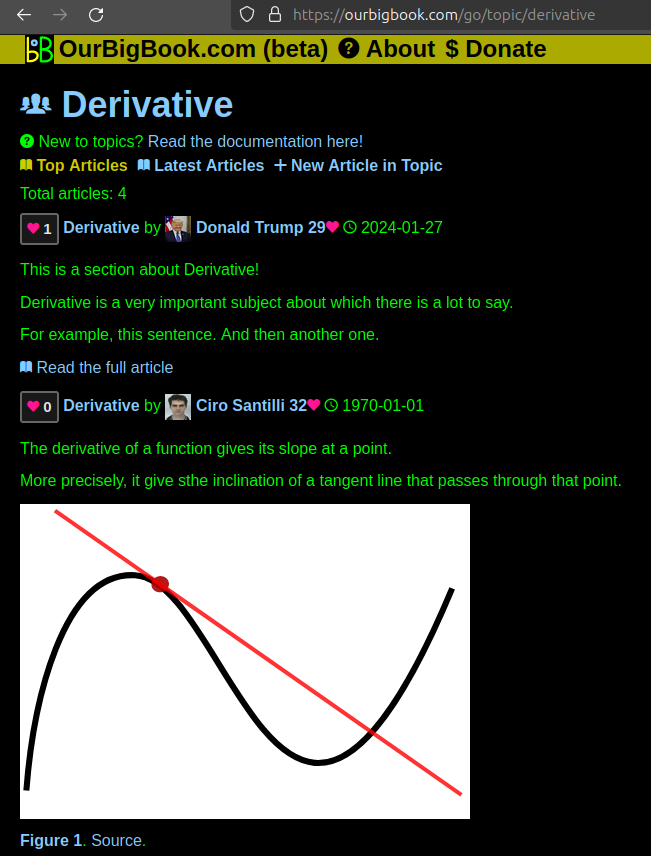
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.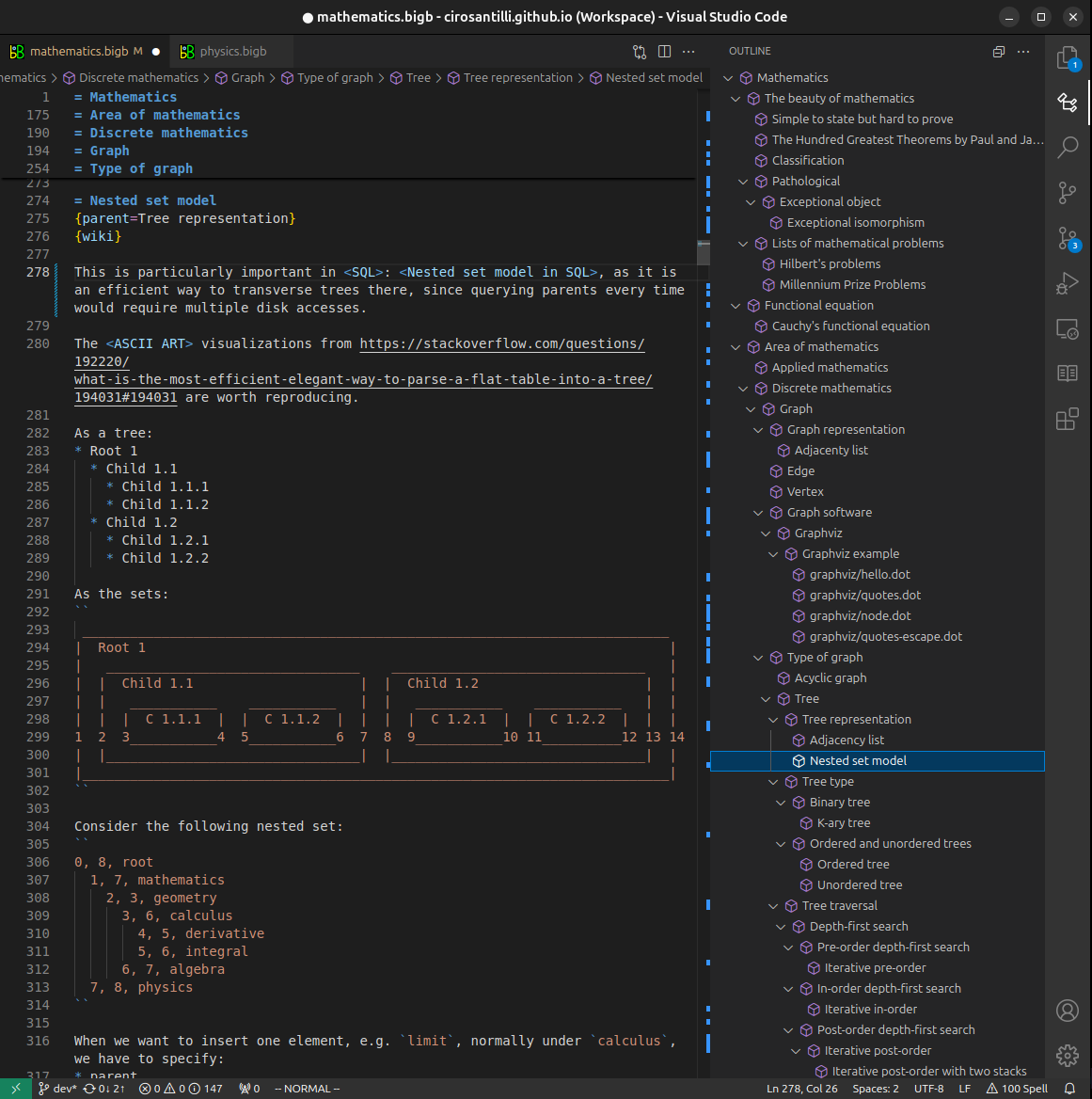
Figure 4. Visual Studio Code extension tree navigation.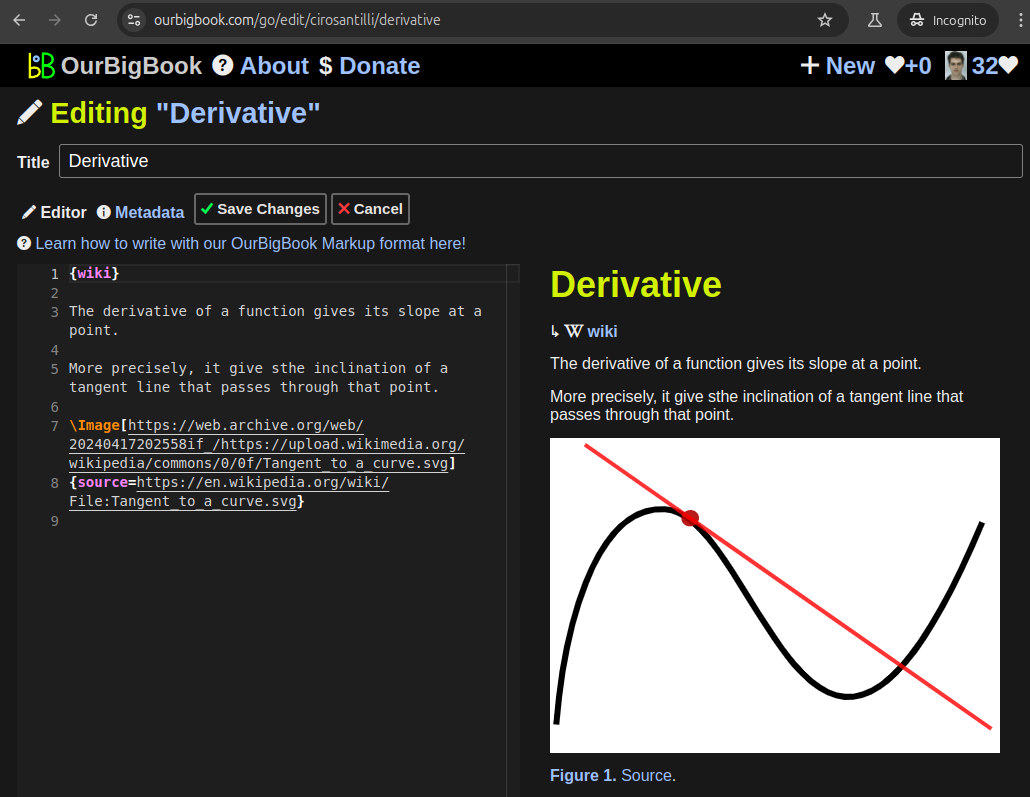
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 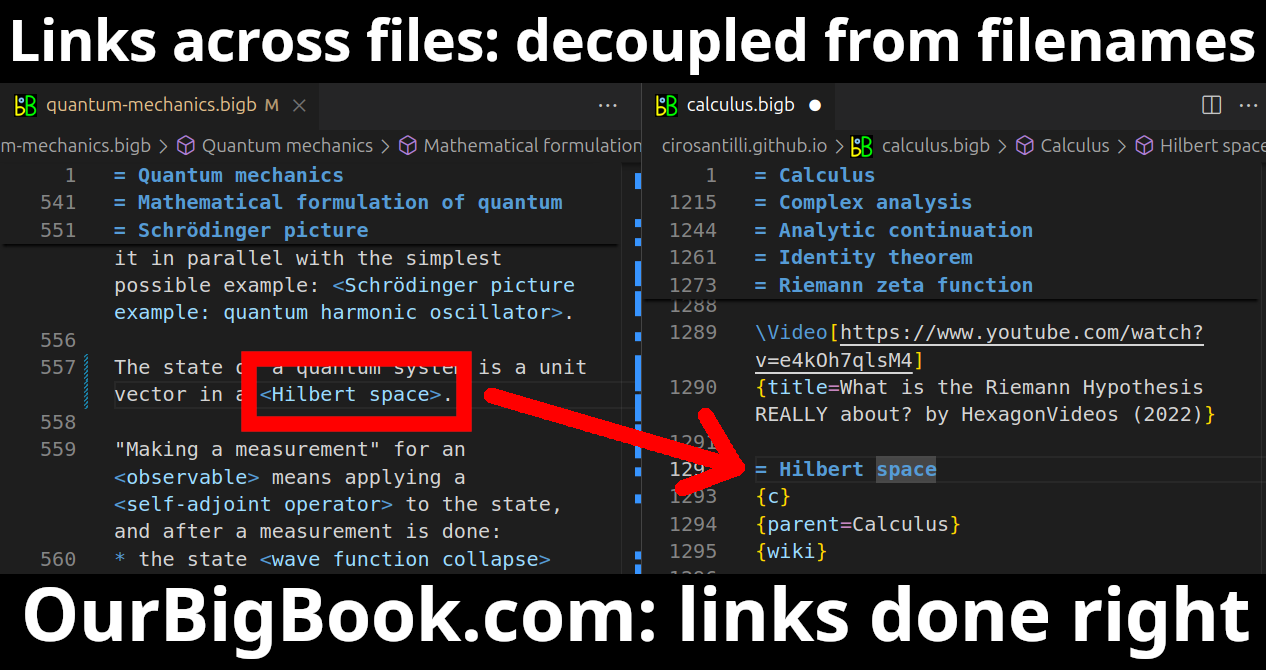
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact