
Incoming links: Molecule
E. Coli K-12 MG1655 gene of unknown function Updated 2025-07-16
E. Coli Whole Cell Model by Covert Lab Source code overview Updated 2025-07-16
Let's try to understand some interesting looking, with a special focus on our understanding of the tiny E. Coli K-12 MG1655 operon thrLABC part of the metabolism, which we have well understood at Section "E. Coli K-12 MG1655 operon thrLABC".
reconstruction/ecoli/flat/compartments.tsvcontains cellular compartment information:"abbrev" "id" "n" "CCO-BAC-NUCLEOID" "j" "CCO-CELL-PROJECTION" "w" "CCO-CW-BAC-NEG" "c" "CCO-CYTOSOL" "e" "CCO-EXTRACELLULAR" "m" "CCO-MEMBRANE" "o" "CCO-OUTER-MEM" "p" "CCO-PERI-BAC" "l" "CCO-PILUS" "i" "CCO-PM-BAC-NEG"CCO: "Celular COmpartment"BAC-NUCLEOID: nucleoidCELL-PROJECTION: cell projectionCW-BAC-NEG: TODO confirm: cell wall (of a Gram-negative bacteria)CYTOSOL: cytosolEXTRACELLULAR: outside the cellMEMBRANE: cell membraneOUTER-MEM: bacterial outer membranePERI-BAC: periplasmPILUS: pilusPM-BAC-NEG: TODO: plasma membrane, but that is the same as cell membrane no?
reconstruction/ecoli/flat/promoters.tsvcontains promoter information. Simple file, sample lines:corresponds to E. Coli K-12 MG1655 promoter thrLp, which starts as position 148."position" "direction" "id" "name" 148 "+" "PM00249" "thrLp"reconstruction/ecoli/flat/proteins.tsvcontains protein information. Sample line corresponding to e. Coli K-12 MG1655 gene thrA:so we understand that:"aaCount" "name" "seq" "comments" "codingRnaSeq" "mw" "location" "rnaId" "id" "geneId" [91, 46, 38, 44, 12, 53, 30, 63, 14, 46, 89, 34, 23, 30, 29, 51, 34, 4, 20, 0, 69] "ThrA" "MRVL..." "Location information from Ecocyc dump." "AUGCGAGUGUUG..." [0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 89103.51099999998, 0.0, 0.0, 0.0, 0.0] ["c"] "EG10998_RNA" "ASPKINIHOMOSERDEHYDROGI-MONOMER" "EG10998"aaCount: amino acid count, how many of each of the 20 proteinogenic amino acid are thereseq: full sequence, using the single letter abbreviation of the proteinogenic amino acidsmw; molecular weight? The 11 components appear to be given atreconstruction/ecoli/flat/scripts/unifyBulkFiles.py:so they simply classify the weight? Presumably this exists for complexes that have multiple classes?molecular_weight_keys = [ '23srRNA', '16srRNA', '5srRNA', 'tRNA', 'mRNA', 'miscRNA', 'protein', 'metabolite', 'water', 'DNA', 'RNA' # nonspecific RNA ]23srRNA,16srRNA,5srRNAare the three structural RNAs present in the ribosome: 23S ribosomal RNA, 16S ribosomal RNA, 5S ribosomal RNA, all others are obvious:- tRNA
- mRNA
- protein. This is the seventh class, and this enzyme only contains mass in this class as expected.
- metabolite
- water
- DNA
- RNA: TODO
rnavsmiscRNA
location: cell compartment where the protein is present,cdefined atreconstruction/ecoli/flat/compartments.tsvas cytoplasm, as expected for something that will make an amino acid
reconstruction/ecoli/flat/rnas.tsv: TODO vstranscriptionUnits.tsv. Sample lines:"halfLife" "name" "seq" "type" "modifiedForms" "monomerId" "comments" "mw" "location" "ntCount" "id" "geneId" "microarray expression" 174.0 "ThrA [RNA]" "AUGCGAGUGUUG..." "mRNA" [] "ASPKINIHOMOSERDEHYDROGI-MONOMER" "" [0.0, 0.0, 0.0, 0.0, 790935.00399999996, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0] ["c"] [553, 615, 692, 603] "EG10998_RNA" "EG10998" 0.0005264904halfLife: half-lifemw: molecular weight, same as inreconstruction/ecoli/flat/proteins.tsv. This molecule only have weight in themRNAclass, as expected, as it just codes for a proteinlocation: same as inreconstruction/ecoli/flat/proteins.tsvntCount: nucleotide count for each of the ATGCmicroarray expression: presumably refers to DNA microarray for gene expression profiling, but what measure exactly?
reconstruction/ecoli/flat/sequence.fasta: FASTA DNA sequence, first two lines:>E. coli K-12 MG1655 U00096.2 (1 to 4639675 = 4639675 bp) AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGreconstruction/ecoli/flat/transcriptionUnits.tsv: transcription units. We can observe for example the two different transcription units of the E. Coli K-12 MG1655 operon thrLABC in the lines:"expression_rate" "direction" "right" "terminator_id" "name" "promoter_id" "degradation_rate" "id" "gene_id" "left" 0.0 "f" 310 ["TERM0-1059"] "thrL" "PM00249" 0.198905992329492 "TU0-42486" ["EG11277"] 148 657.057317358791 "f" 5022 ["TERM_WC-2174"] "thrLABC" "PM00249" 0.231049060186648 "TU00178" ["EG10998", "EG10999", "EG11000", "EG11277"] 148promoter_id: matches promoter id inreconstruction/ecoli/flat/promoters.tsvgene_id: matches id inreconstruction/ecoli/flat/genes.tsvid: matches exactly those used in BioCyc, which is quite nice, might be more or less standardized:
reconstruction/ecoli/flat/genes.tsv"length" "name" "seq" "rnaId" "coordinate" "direction" "symbol" "type" "id" "monomerId" 66 "thr operon leader peptide" "ATGAAACGCATT..." "EG11277_RNA" 189 "+" "thrL" "mRNA" "EG11277" "EG11277-MONOMER" 2463 "ThrA" "ATGCGAGTGTTG" "EG10998_RNA" 336 "+" "thrA" "mRNA" "EG10998" "ASPKINIHOMOSERDEHYDROGI-MONOMER"reconstruction/ecoli/flat/metabolites.tsvcontains metabolite information. Sample lines:In the case of the enzyme thrA, one of the two reactions it catalyzes is "L-aspartate 4-semialdehyde" into "Homoserine"."id" "mw7.2" "location" "HOMO-SER" 119.12 ["n", "j", "w", "c", "e", "m", "o", "p", "l", "i"] "L-ASPARTATE-SEMIALDEHYDE" 117.104 ["n", "j", "w", "c", "e", "m", "o", "p", "l", "i"]Starting from the enzyme page: biocyc.org/gene?orgid=ECOLI&id=EG10998 we reach the reaction page: biocyc.org/ECOLI/NEW-IMAGE?type=REACTION&object=HOMOSERDEHYDROG-RXN which has reaction IDHOMOSERDEHYDROG-RXN, and that page which clarifies the IDs:so these are the compounds that we care about.- biocyc.org/compound?orgid=ECOLI&id=L-ASPARTATE-SEMIALDEHYDE: "L-aspartate 4-semialdehyde" has ID
L-ASPARTATE-SEMIALDEHYDE - biocyc.org/compound?orgid=ECOLI&id=HOMO-SER: "Homoserine" has ID
HOMO-SER
- biocyc.org/compound?orgid=ECOLI&id=L-ASPARTATE-SEMIALDEHYDE: "L-aspartate 4-semialdehyde" has ID
reconstruction/ecoli/flat/reactions.tsvcontains chemical reaction information. Sample lines:"reaction id" "stoichiometry" "is reversible" "catalyzed by" "HOMOSERDEHYDROG-RXN-HOMO-SER/NAD//L-ASPARTATE-SEMIALDEHYDE/NADH/PROTON.51." {"NADH[c]": -1, "PROTON[c]": -1, "HOMO-SER[c]": 1, "L-ASPARTATE-SEMIALDEHYDE[c]": -1, "NAD[c]": 1} false ["ASPKINIIHOMOSERDEHYDROGII-CPLX", "ASPKINIHOMOSERDEHYDROGI-CPLX"] "HOMOSERDEHYDROG-RXN-HOMO-SER/NADP//L-ASPARTATE-SEMIALDEHYDE/NADPH/PROTON.53." {"NADPH[c]": -1, "NADP[c]": 1, "PROTON[c]": -1, "L-ASPARTATE-SEMIALDEHYDE[c]": -1, "HOMO-SER[c]": 1 false ["ASPKINIIHOMOSERDEHYDROGII-CPLX", "ASPKINIHOMOSERDEHYDROGI-CPLX"]catalized by: here we seeASPKINIHOMOSERDEHYDROGI-CPLX, which we can guess is a protein complex made out ofASPKINIHOMOSERDEHYDROGI-MONOMER, which is the ID for thethrAwe care about! This is confirmed incomplexationReactions.tsv.
reconstruction/ecoli/flat/complexationReactions.tsvcontains information about chemical reactions that produce protein complexes:The"process" "stoichiometry" "id" "dir" "complexation" [ { "molecule": "ASPKINIHOMOSERDEHYDROGI-CPLX", "coeff": 1, "type": "proteincomplex", "location": "c", "form": "mature" }, { "molecule": "ASPKINIHOMOSERDEHYDROGI-MONOMER", "coeff": -4, "type": "proteinmonomer", "location": "c", "form": "mature" } ] "ASPKINIHOMOSERDEHYDROGI-CPLX_RXN" 1coeffis how many monomers need to get together for form the final complex. This can be seen from the Summary section of ecocyc.org/gene?orgid=ECOLI&id=ASPKINIHOMOSERDEHYDROGI-MONOMER:Fantastic literature summary! Can't find that in database form there however.Aspartate kinase I / homoserine dehydrogenase I comprises a dimer of ThrA dimers. Although the dimeric form is catalytically active, the binding equilibrium dramatically favors the tetrameric form. The aspartate kinase and homoserine dehydrogenase activities of each ThrA monomer are catalyzed by independent domains connected by a linker region.
reconstruction/ecoli/flat/proteinComplexes.tsvcontains protein complex information:"name" "comments" "mw" "location" "reactionId" "id" "aspartate kinase / homoserine dehydrogenase" "" [0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 356414.04399999994, 0.0, 0.0, 0.0, 0.0] ["c"] "ASPKINIHOMOSERDEHYDROGI-CPLX_RXN" "ASPKINIHOMOSERDEHYDROGI-CPLX"reconstruction/ecoli/flat/protein_half_lives.tsvcontains the half-life of proteins. Very few proteins are listed however for some reason.reconstruction/ecoli/flat/tfIds.csv: transcription factors information:"TF" "geneId" "oneComponentId" "twoComponentId" "nonMetaboliteBindingId" "activeId" "notes" "arcA" "EG10061" "PHOSPHO-ARCA" "PHOSPHO-ARCA" "fnr" "EG10325" "FNR-4FE-4S-CPLX" "FNR-4FE-4S-CPLX" "dksA" "EG10230"
Molecular beam Created 2024-11-15 Updated 2025-07-16
Molecular beams are cool because they create a one dimensional flow of molecules, which makes it easier to observe certain single-molecule effects, as it removes the multi-particle issues from experiments.
Key molecular beam experiments include:
- Stern-Gerlach experiment, which confirmed the existence of spin
- Rabi's NMR experiment, which confirmed the existence of nuclear spin
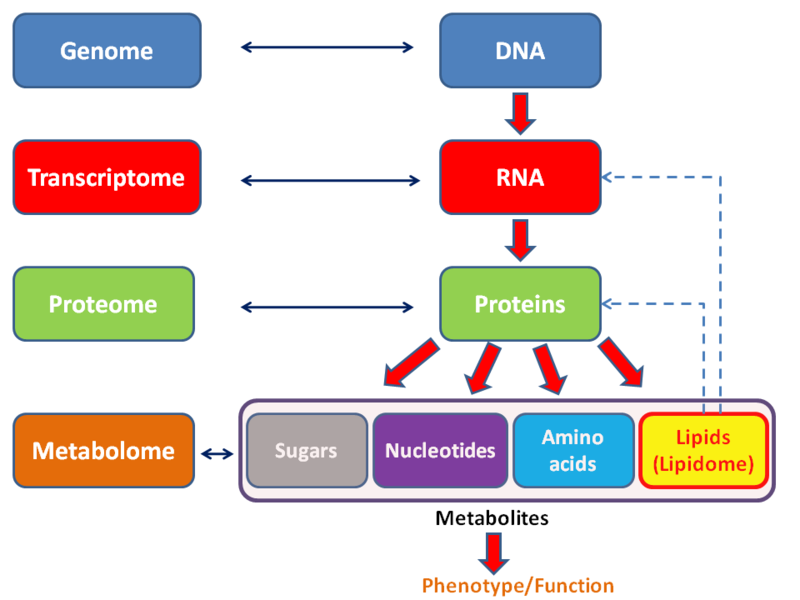
Omics Updated 2025-07-16
Each of the omics studies a subset of molecular biology with a data intensive and broad point of view that tries to understand global function or organisms, trying to understand what every biologically relevant molecule does as part of the hole metabolism.
The main omics are:
Omics might be stamp collecting, but maybe it is a bit more like Trading card game/Magic: The Gathering collecting, in which the cards that you are collecting actually have specific uses and interactions, especially considering that most metabolic pathways are analogous across many species.
Quantum chemistry Updated 2025-07-16
Ah, the jewel of computational physics.
Also known as an ab initio method: no experimental measurement is taken as input, QED is all you need.
But since QED is thought to fully describe all relevant aspects molecules, it could be called "the" ab initio method.
For one, if we were able to predict protein molecule interactions, our understanding of molecular biology technologies would be solved.
No more ultra expensive and complicated X-ray crystallography or cryogenic electron microscopy.
And the fact that quantum computers are one of the most promising advances to this field, is also very very exciting: Section "Quantum algorithm".