Thrust is a fundamental concept in physics and engineering, particularly in the fields of mechanics and aerodynamics. It refers to the force that propels an object forward, typically generated by engines or motors.
Tractive force refers to the force exerted by a vehicle or machine to pull or move a load. It is a crucial concept in the fields of mechanics, transportation, and engineering, particularly in the context of vehicles such as trains, trucks, or agricultural machinery. The tractive force is essential for overcoming resistive forces such as friction, gradient (slope), and inertia.
The list of minor planets numbered between 28001 and 29000 includes various small celestial bodies that are primarily located in the asteroid belt between Mars and Jupiter. These minor planets can consist of asteroids, comets, and other small objects in the solar system. The complete list of minor planets in this range would typically include details such as their discovery dates, names, and other relevant information.
The List of minor planets from 40001 to 41000 includes various celestial objects that are classified as minor planets, or asteroids, within the asteroid belt or beyond. These minor planets are typically designated with a number and may also have a name or designation based on mythology, geography, or notable figures.
The list of minor planets from 607001 to 608000 includes various small celestial bodies in our solar system that have been officially designated with a numeric identifier by the International Astronomical Union (IAU). These minor planets can include asteroids, centaurs, and other small Solar System bodies that are not classified as comets or planets.
The "List of minor planets: 75001–76000" refers to a specific segment of minor planets (or asteroids) that have been assigned numbers in the range of 75001 to 76000 by the International Astronomical Union (IAU). These numbers are part of a larger catalog of minor planets, which are small celestial bodies orbiting the Sun, primarily located in the asteroid belt between Mars and Jupiter.
A list of firearms typically refers to a comprehensive catalog or collection of different types of firearms, which can include categories such as handguns, rifles, shotguns, and machine guns. These lists can provide various details about each firearm, including: 1. **Type of Firearm**: Classification such as pistol, revolver, rifle, shotgun, etc. 2. **Manufacturer**: The company or brand that produces the firearm.
Logarithmic decrement is a measure used in the field of vibration analysis and control to quantify the rate of decay of oscillations in a damped system. It is particularly useful in systems that exhibit harmonic motion, such as mechanical systems with damping (e.g., springs, beams, and electrical circuits). The logarithmic decrement (\(\delta\)) is defined as the natural logarithm of the ratio of the amplitudes of two successive peaks in the decay of oscillation.
Louis Mink is a notable figure primarily known for his contributions to the philosophy of history and narrative theory. He is particularly recognized for his views on how historical events are understood and represented through narratives. Mink argued that history is not just a straightforward account of events but is shaped by the interpretations and stories that historians create around those events. His work emphasizes the importance of narrative structure in understanding history and the ways in which different interpretations can influence our perception of the past.
Low surface brightness (LSB) galaxies are a type of galaxy characterized by their relatively low luminosity per unit area. This means that they are fainter and have less concentrated light compared to more conventional, high surface brightness galaxies (HSB). Here are some key features and characteristics of LSB galaxies: 1. **Surface Brightness**: LSB galaxies have a surface brightness that is significantly lower than that of typical galaxies.
Łukasiewicz logic, named after the Polish logician Jan Łukasiewicz, is a non-classical system of logic that extends classical propositional logic. It is particularly known for its treatment of many-valued logics, where it allows for more than just the binary true and false values. 1. **Many-Valued Logic**: One of the key contributions of Łukasiewicz is his development of many-valued logic, which allows for a spectrum of truth values.
Macintosh operating systems development refers to the process of creating, evolving, and maintaining the macOS (formerly known as Mac OS X and OS X) operating system used by Apple's line of Macintosh computers. This development encompasses various aspects, including architecture, design, features, user interface, performance optimization, security, and application compatibility. ### Historical Context: 1. **Early Beginnings (1984)**: The original Macintosh operating system was launched alongside the first Macintosh computer in 1984.
The Malus-Dupin theorem, also known simply as Malus's law, is a principle in optics that describes the intensity of polarized light as it passes through a polarizing filter. It states that the intensity of light transmitted through a polarizer is proportional to the square of the cosine of the angle between the light's initial polarization direction and the axis of the polarizer.
The Mark I Fire Control Computer is a historical computing device used by the United States Navy from the early to mid-20th century. Developed during World War II, it was one of the earliest examples of a digital computer and was specifically designed to assist in naval artillery fire control.
Double recursion refers to a recursive function that makes two recursive calls within its body, rather than just one. This technique can be found in various algorithms, particularly in problems involving tree structures, combinatorial calculations, or when dealing with problems that can be broken down into multiple subproblems. A classic example of double recursion is the computation of the Fibonacci sequence.
The Mason-Dixon line is a survey boundary that was originally established in the 18th century to resolve a border dispute between the British colonies of Maryland and Pennsylvania. Surveyors Charles Mason and Jeremiah Dixon carried out the survey from 1763 to 1767. The line runs approximately along the latitude of 39°43′ N and came to symbolize the cultural division between the Northern and Southern U.S. states, particularly during the period leading up to the Civil War.
Trailing Twelve Months (TTM) is a financial metric that measures a company's performance over the most recent 12-month period. It is commonly used in various financial analyses to assess a company's revenue, earnings, or other performance indicators, and it helps analysts and investors to get a more current view of the company's financial health compared to traditional annual reports.
A live birth in humans refers to the successful delivery of a baby who shows signs of life after birth, such as breathing, heartbeat, or voluntary muscle movement. This term is typically used in medical and demographic contexts to distinguish between live births and stillbirths, where the fetus has died in utero before or during delivery. Live births are an important measure in public health and statistics, as they indicate successful pregnancies and the health of mothers and infants.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.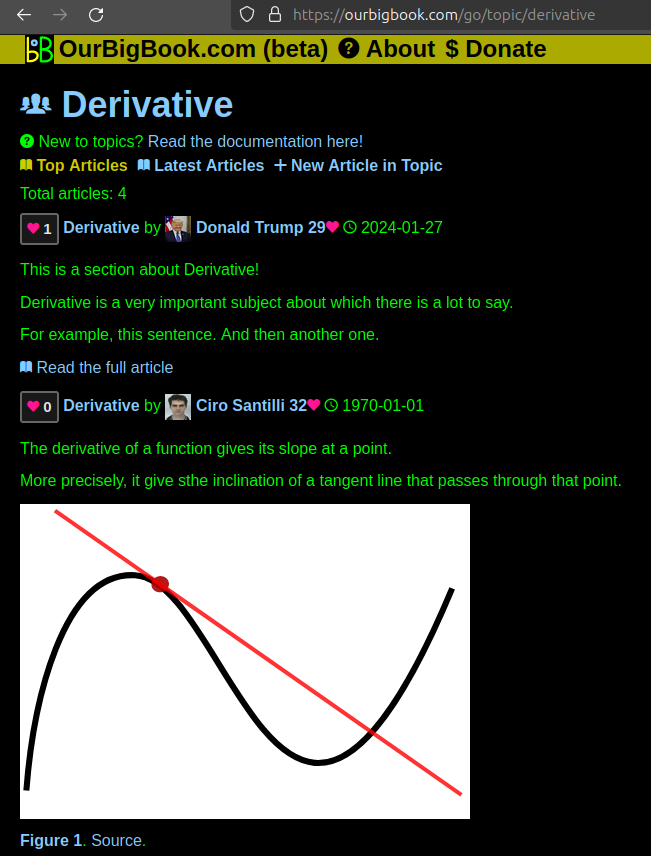
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.
Figure 4. Visual Studio Code extension tree navigation.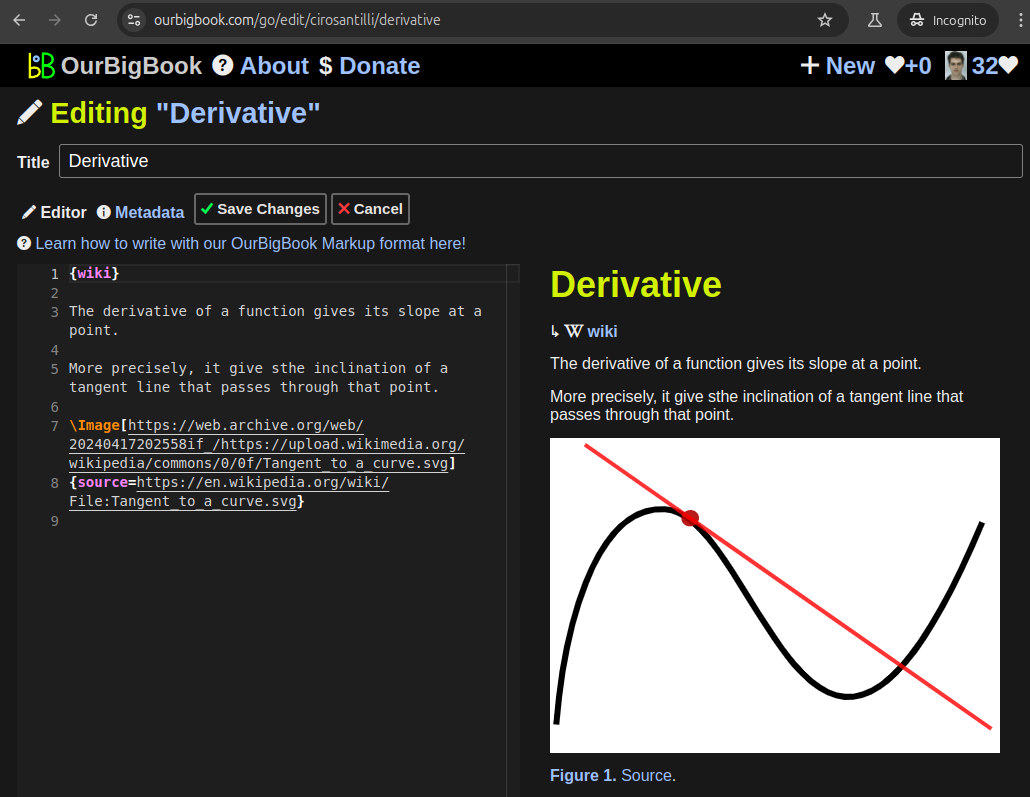
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 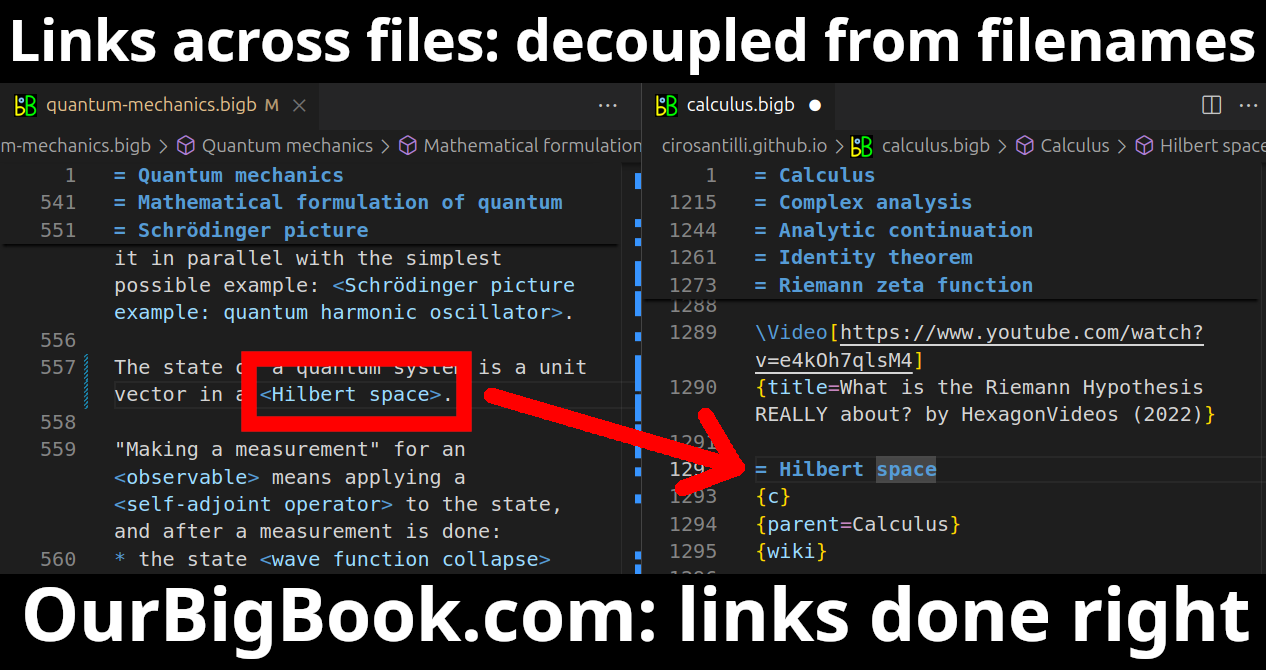
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact