Kitchen: 2021-04: nine-volt battery battery at 8.79V. Not sounding.
Haven't found the one yet:
- open source software, doh
- end-to-end encryption...
- has browser frontend and Android app
- public URL without sharing your mobile phone: messaging software that force you to have a mobile phone
- self-destroying messages (turned on by default please)
- user base large enough to give some confidence that it was reviewed for security issues
- easy/built-in setup over Tor
Optional but really ideal:
- can delete messages from the device of the person you sent it to, no matter how old
- decentralized, your username is a public key
The state of messaging is ridiculous as of 2020.
Programming languages without a decent dominating package system by  Ciro Santilli 40 Updated 2025-07-16
Ciro Santilli 40 Updated 2025-07-16
Ciro Santilli 40 Updated 2025-07-16Largest programs ever written:
It is interesting to note how late C appeared: 1972, compared e.g. to Fortran which is from 1957. This is basically because C was a "systems programming language", i.e. with focus on pointer manipulation, and because early computers were so weak, there was no operating system or many software layers in the early days. Fortran however was a numerical language, and it ran directly on bare metal, an application that existed before systems programming.
Examples under c.
The key question is: why is this not symmetrical?
One answer is: because one of the twin accelerates, and therefore changes inertial frames.
But the better answer is: understand what happens when the stationary twin sends light signals at constant time intervals to each other. When does the travelling twin receives them?

Another way of understanding it is: you have to make all calculations on a single inertial frame for the entire trip.
Supposing the sibling quickly accelerates out (or magically starts moving at constant speed), travels at constant speed, and quickly accelerates back, and travels at constant speed setup, there are three frames that seem reasonable:
- the frame of the non-accelerating sibling
- the outgoing trip of the accelerating sibling
- the return trip of the accelerating sibling
If you do that, all three calculations give the exact same result, which is reassuring.
Another way to understand it is to do explicit integrations of the acceleration: physics.stackexchange.com/questions/242043/what-is-the-proper-way-to-explain-the-twin-paradox/242044#242044 This is the least insightful however :-)
Bibliography:
Ciro Santilli managed to port it to Sequelize for PostgreSQL as shown at: github.com/cirosantilli/feathers-chat/tree/sequelize-pg
Amazingly confirms the wave particle duality of quantum mechanics.
The effect is even more remarkable when done with individual particles such individual photons or electrons.
Richard Feynman liked to stress how this experiment can illustrate the core ideas of quantum mechanics. Notably, he night have created the infinitely many slits thought experiment which illustrates the path integral formulation.
Ubuntu 23.04 install:
sudo apt install rbaseHello world:
R -e 'print("hello world")'Explaining this was was one of the key initial achievements of the Dirac equation.
Yes, but this is not predicted by the Schrödinger equation, you need to go to the Dirac equation.
See also:
- physics.stackexchange.com/questions/233330/why-do-electrons-jump-between-orbitals
- physics.stackexchange.com/questions/117417/quantum-mechanics-scattering-theory/522220#522220
- physics.stackexchange.com/questions/430268/stimulated-emission-how-can-giving-energy-to-electrons-make-them-decay-to-a-low/430288
TODO understand better, mentioned e.g. at Subtle is the Lord by Abraham Pais (1982) page 20, and is something that Einstein worked on.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.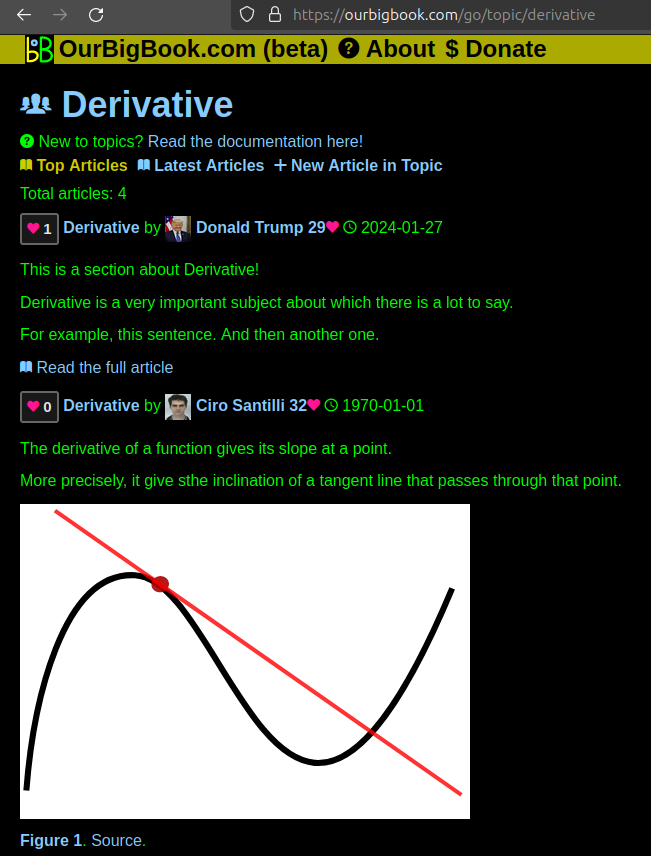
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
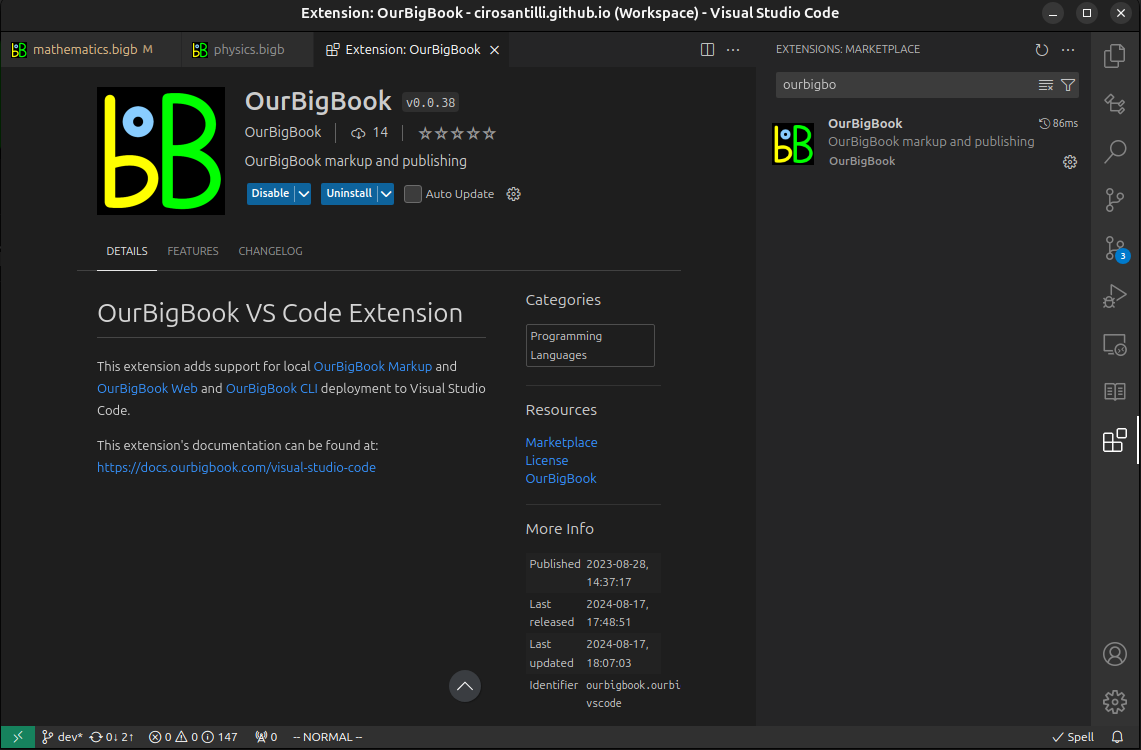
Figure 3. Visual Studio Code extension installation.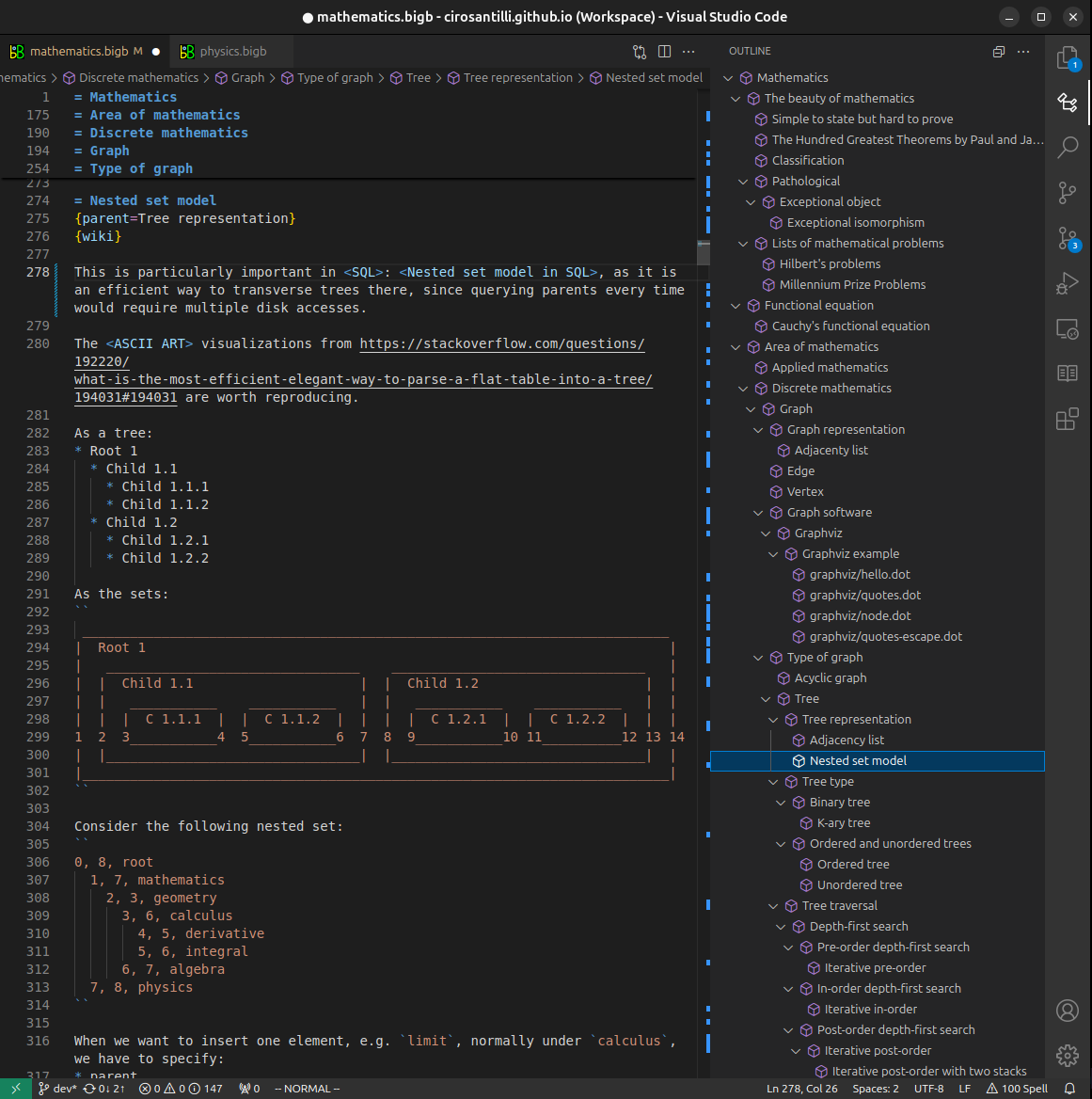
Figure 4. Visual Studio Code extension tree navigation.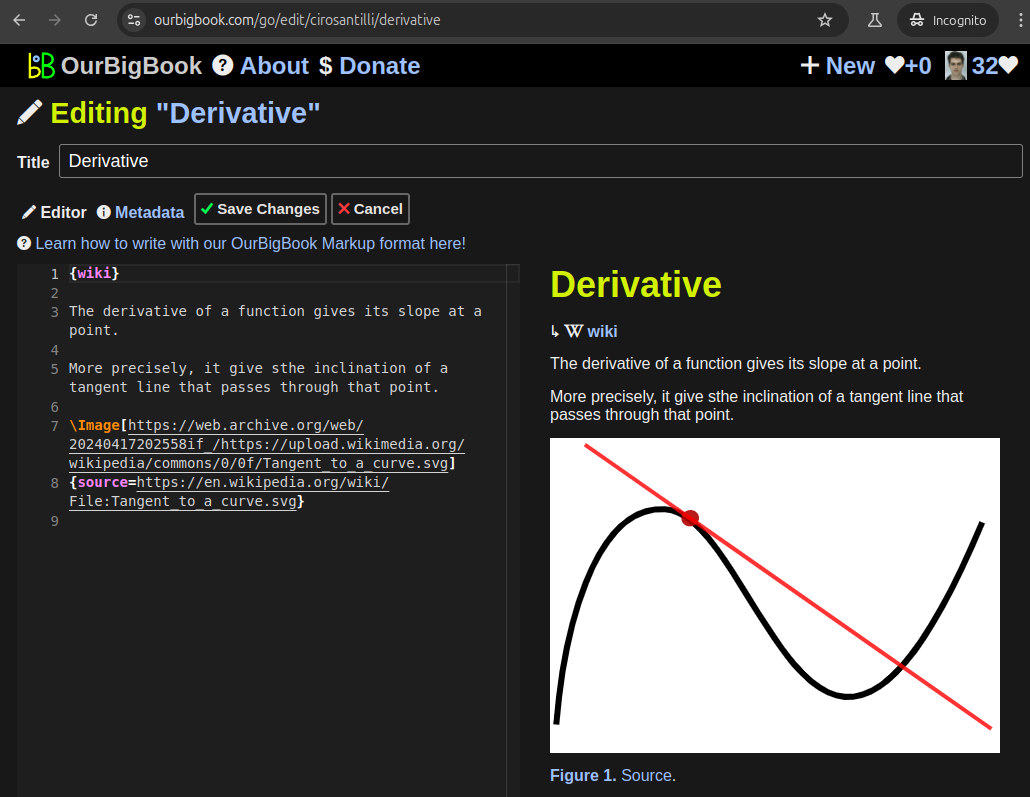
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 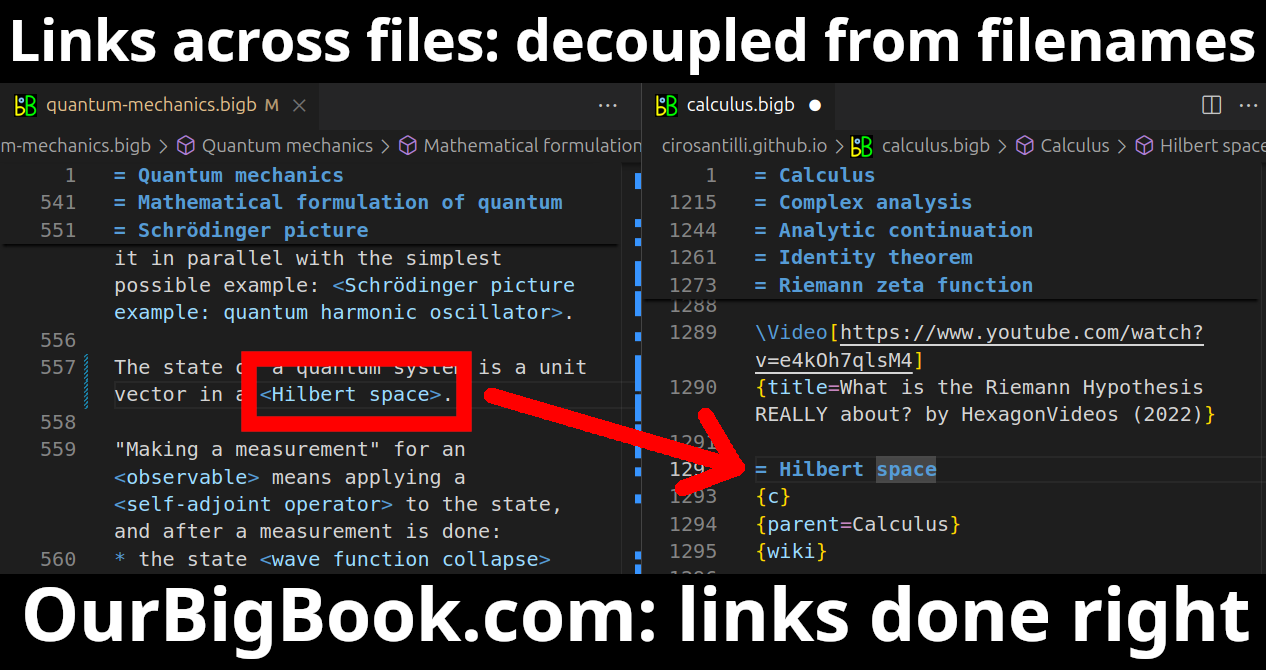
- Infinitely deep tables of contents:


{kind=link}
All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact