The 1940s was a pivotal decade for robotics, as it laid the groundwork for future developments in automation and robotic technology. Here are some key highlights from that period: 1. **Early Concepts**: The term "robot" was popularized by Karel Čapek's 1920 play "R.U.R." (Rossum's Universal Robots), which introduced the idea of artificial beings created to serve humans. This concept spurred interest in the potential of machines to perform tasks.
The year 2010 was significant in the field of robotics for several reasons, including advancements in technology, notable events, and the introduction of influential robotics platforms and competitions. Here are some key highlights from 2010 in robotics: 1. **Robotics Competitions**: Various robotics competitions took place in 2010, including the RoboCup and FIRST Robotics Competition. These events promoted innovation and collaboration among students and professionals in the field.
TX-2 can refer to a few different things, depending on the context. Here are a couple of notable references: 1. **TX-2 (Computer System)**: TX-2 was an early experimental computer developed in the 1950s at the Massachusetts Institute of Technology (MIT). It was an advanced machine for its time, featuring innovations like multitasking and the use of high-level programming languages.
Thinning in the context of mathematical morphology is a morphological operation used primarily in image processing and computer vision. It is a technique that reduces the thickness of objects in a binary image while preserving their connectivity and shape. The goal of thinning is to simplify the representation of features in an image, often used for tasks like shape analysis, object recognition, or preprocessing for further analysis.
The Remington Rand 409 refers to a model of typewriter produced by the Remington Rand company, which was a significant manufacturer of typewriters and office equipment in the 20th century. The Remington Rand 409 is known for its portable design and durability, characteristic of many typewriters produced during that era.
NYIT Bears lacrosse refers to the lacrosse team representing the New York Institute of Technology (NYIT), which is located in Old Westbury, New York. The team is part of NCAA Division II and competes in the East Coast Conference (ECC). The NYIT Bears lacrosse program has a history of participation in collegiate lacrosse, striving for excellence both on the field and academically. The team has been known for its commitment to developing players' skills and promoting teamwork and sportsmanship.
The Reeds–Sloane algorithm is an approach in computer science, specifically in the field of algorithm design and geometric optimization. It provides a way to find the shortest path or the optimal sequence of operations for navigating a search space, often applied in problems related to robotics and motion planning. The algorithm is particularly notable for its application in situations where movements are constrained to a fixed set of directions or within a grid-like structure.
SC2000, also known as Schematic Capture 2000, is a software tool commonly used for electronic design automation (EDA). Specifically, it focuses on the schematic capture phase of circuit design, allowing engineers and designers to create and manage electronic schematics. The software may support functionalities such as simulation, layout, and design rule checking, making it easier to design and validate electronic circuits before moving to the physical layout stage.
The Subject Alternative Name (SAN) is an extension to the X.509 specification that allows users to specify additional host names for a single SSL certificate. It was introduced to avoid the limitations of the Common Name (CN) field in SSL certificates. The SAN field can include multiple values, which may consist of: 1. **DNS Names**: Additional domain names (e.g., www.example.com, example.org). 2. **IP Addresses**: Specific IP addresses associated with the certificate.
Disk encryption theory refers to the principles and techniques used to protect data stored on physical storage devices, like hard drives or SSDs, by converting it into a format that cannot be read without proper authorization. The main goals of disk encryption are to maintain data confidentiality, prevent unauthorized access, and protect sensitive information from theft or loss.
World War II Japanese cryptography refers to the cryptographic methods and systems used by Japan during World War II for secure communication. The Japanese military and government employed various techniques to encode and decode messages, some of which were highly sophisticated. Key aspects of Japanese cryptography during this period include: 1. **Cipher Machines**: The Japanese used several cipher machines, the most notable being the **Purple machine** (JN-25). This machine was an electro-mechanical device used to encrypt diplomatic messages.
German submarine U-505 is one of the most famous U-boats from World War II. It was a Type IXC U-boat of the Kriegsmarine (German Navy) and was notable for being the first U-boat captured by the United States Navy on the high seas.
An SSHFP (SSH Fingerprint) record is a type of DNS (Domain Name System) resource record that provides a way to associate SSH (Secure Shell) public keys with domain names. It allows clients connecting to an SSH server to validate the server's identity and verify that they are connecting to the actual server they intend to reach, thereby helping to prevent man-in-the-middle attacks.
NSA cryptography refers to the cryptographic standards, practices, and technologies developed or endorsed by the National Security Agency (NSA) of the United States. The NSA is responsible for monitoring and securing U.S. communications and information systems, and cryptography plays a critical role in these efforts. Key aspects of NSA cryptography include: 1. **Standards Development**: The NSA contributes to the development of cryptographic standards used by the U.S. government, which often influence broader industry standards.
An Information Exchange Gateway (IEG) is a platform or system that facilitates the seamless exchange of data and information between different systems, applications, or organizations. It is often used in contexts where disparate systems need to communicate with each other, ensuring that data can flow freely and securely across different environments. ### Key Features of Information Exchange Gateways: 1. **Interoperability**: IEGs help different software applications and systems that use various protocols or data formats to communicate with each other.
The Spatial Geodesy Research Group typically refers to a research group or academic department focusing on the study of geodesy, which is the science of measuring and understanding the Earth's geometric shape, orientation in space, and gravitational field. Geodesy is essential for applications in navigation, mapping, Earth observation, and understanding tectonic processes.
Sfera is a series of Russian Earth observation satellites. The series is designed to enhance remote sensing capabilities, providing high-resolution imagery and data to support various applications such as agriculture, forestry, environmental monitoring, urban planning, and disaster management. The Sfera satellites are part of Russia's efforts to modernize and expand its satellite capabilities in response to both domestic needs and international demand for Earth observation data.
GPS satellites are part of the Global Positioning System (GPS), a satellite-based navigation system that enables users to determine their exact location (latitude, longitude, and altitude) anywhere on Earth. The system consists of a constellation of satellites that continuously transmit signals to GPS receivers, which can interpret these signals to calculate precise positioning. ### Key Features of GPS Satellites: 1. **Constellation**: The GPS system typically consists of at least 24 operational satellites orbiting the Earth in six orbital planes.
Pinned article: Introduction to the OurBigBook Project
Welcome to the OurBigBook Project! Our goal is to create the perfect publishing platform for STEM subjects, and get university-level students to write the best free STEM tutorials ever.
Everyone is welcome to create an account and play with the site: ourbigbook.com/go/register. We belive that students themselves can write amazing tutorials, but teachers are welcome too. You can write about anything you want, it doesn't have to be STEM or even educational. Silly test content is very welcome and you won't be penalized in any way. Just keep it legal!
Intro to OurBigBook
. Source. We have two killer features:
- topics: topics group articles by different users with the same title, e.g. here is the topic for the "Fundamental Theorem of Calculus" ourbigbook.com/go/topic/fundamental-theorem-of-calculusArticles of different users are sorted by upvote within each article page. This feature is a bit like:
- a Wikipedia where each user can have their own version of each article
- a Q&A website like Stack Overflow, where multiple people can give their views on a given topic, and the best ones are sorted by upvote. Except you don't need to wait for someone to ask first, and any topic goes, no matter how narrow or broad
This feature makes it possible for readers to find better explanations of any topic created by other writers. And it allows writers to create an explanation in a place that readers might actually find it.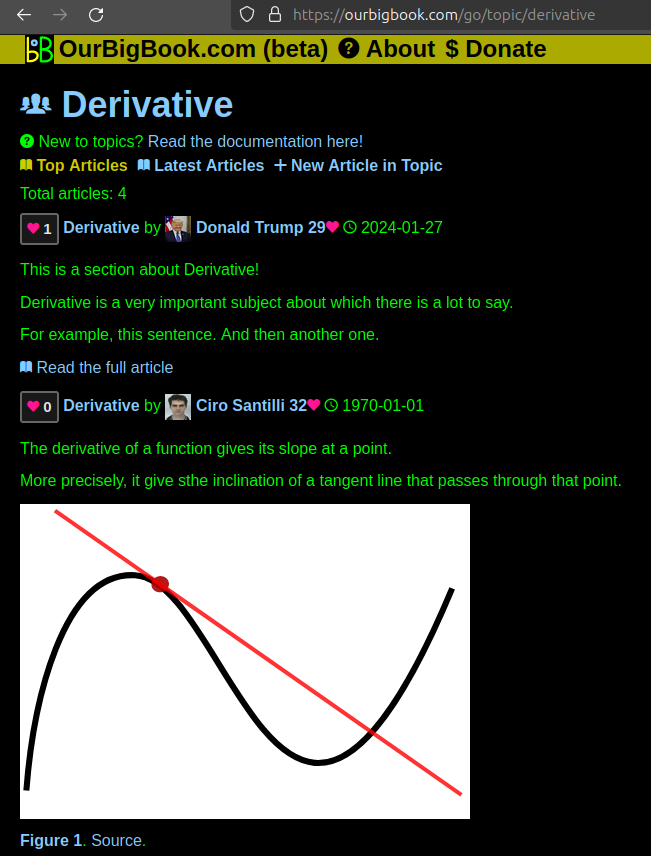
Figure 1. Screenshot of the "Derivative" topic page. View it live at: ourbigbook.com/go/topic/derivativeVideo 2. OurBigBook Web topics demo. Source. - local editing: you can store all your personal knowledge base content locally in a plaintext markup format that can be edited locally and published either:This way you can be sure that even if OurBigBook.com were to go down one day (which we have no plans to do as it is quite cheap to host!), your content will still be perfectly readable as a static site.
- to OurBigBook.com to get awesome multi-user features like topics and likes
- as HTML files to a static website, which you can host yourself for free on many external providers like GitHub Pages, and remain in full control
Figure 3. Visual Studio Code extension installation.
Figure 4. Visual Studio Code extension tree navigation.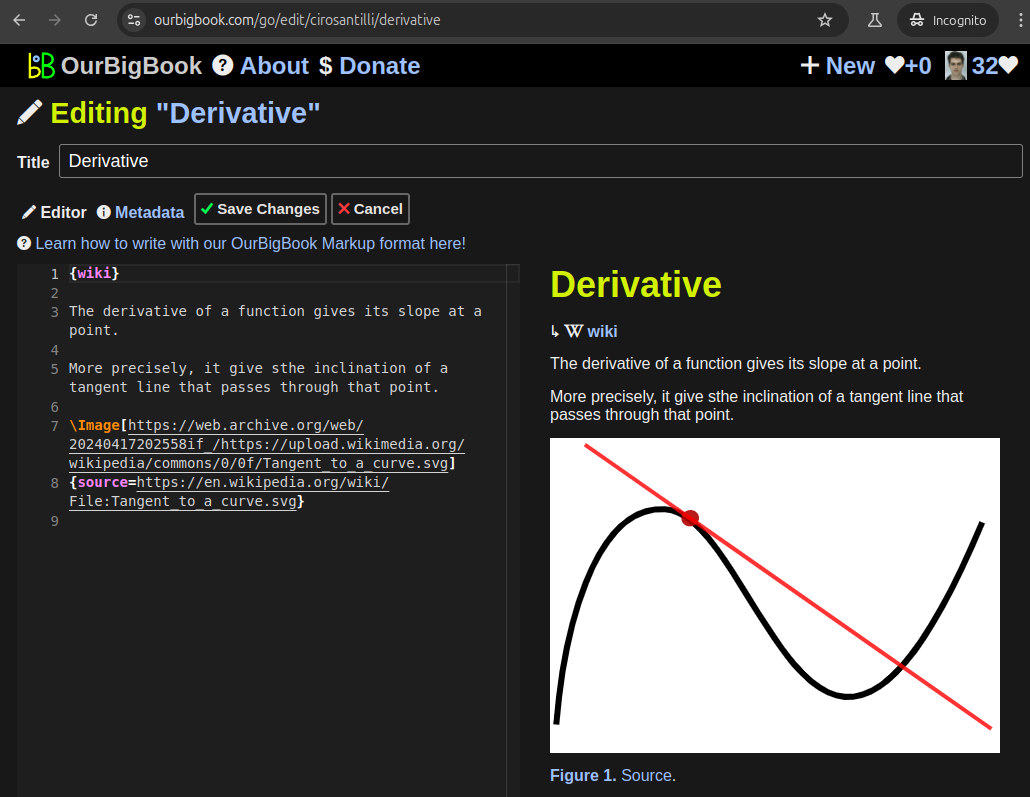
Figure 5. Web editor. You can also edit articles on the Web editor without installing anything locally.Video 3. Edit locally and publish demo. Source. This shows editing OurBigBook Markup and publishing it using the Visual Studio Code extension.Video 4. OurBigBook Visual Studio Code extension editing and navigation demo. Source. 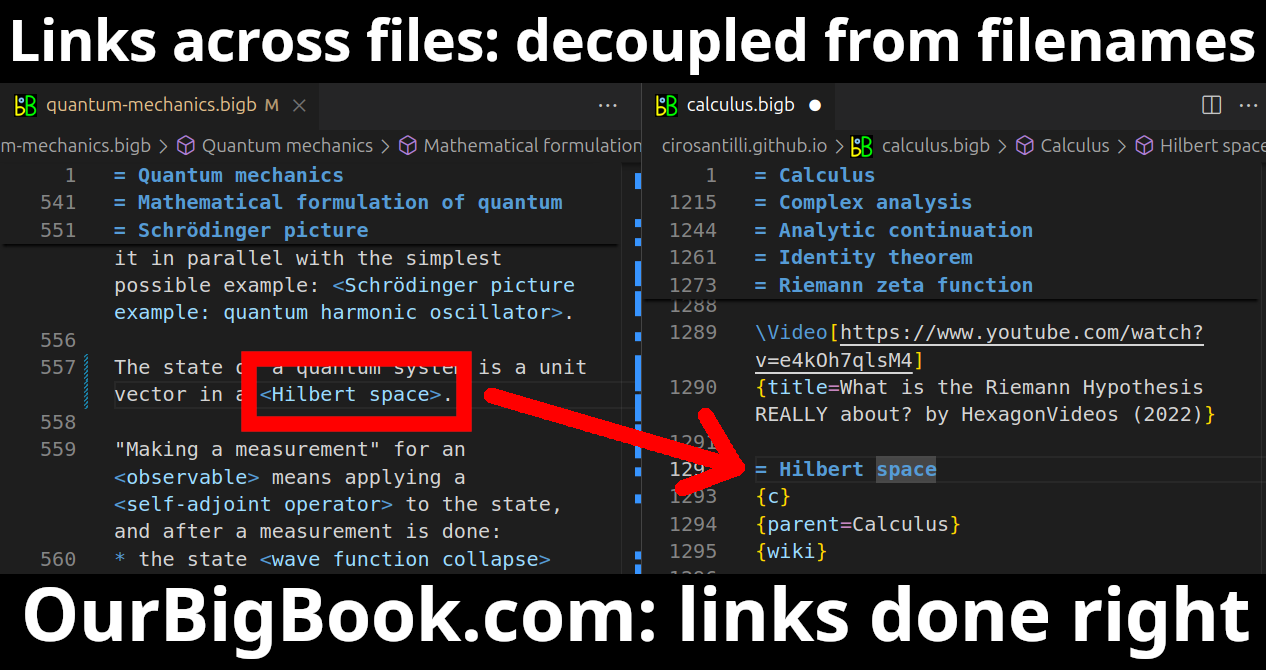
- Infinitely deep tables of contents:


All our software is open source and hosted at: github.com/ourbigbook/ourbigbook
Further documentation can be found at: docs.ourbigbook.com
Feel free to reach our to us for any help or suggestions: docs.ourbigbook.com/#contact