
Incoming links: X-ray crystallography
1914 Nobel Prize in Physics Updated 2025-07-16
Not only did this open the way for X-ray crystallography, it more fundamentally clarified the nature of X-rays as being electromagnetic radiation, and helped further establish the atomic theory.
1962 Nobel Prize in Chemistry Updated 2025-07-16
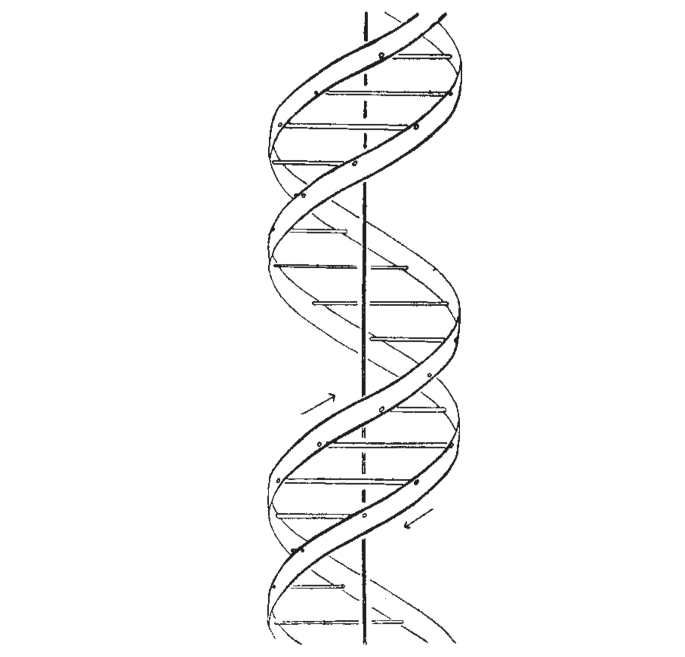
A Structure for Deoxyribose Nucleic Acid Created 2025-06-12 Updated 2025-07-16
Starting line:The Eighth Day of Creation explains the "salt" part as that was the usual way to prepare DNA for X-ray crystallography, where something binds with the phosphate groups of DNA
We wish to suggest a structure for the salt of deoxyribose nucleic acid (D.N.A,). This structure has novel features which are of considerable biological interest.
The paper then shoots down other previously devised helical structures, notably some containing 3 strands or phosphate on the inside.
Then they briefly describe their structure, and promise more details on future articles. This was mostly a short one-page priority note.
Then they drop their shell bomb conclusion:
It has not es~aped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material.
Both Wilkins and Rosalind Franklin are acknowledged at the end.
Atom Updated 2025-07-16
Much before atoms were thought to be "experimentally real", chemists from the 19th century already used "conceptual atoms" as units for the proportions observed in macroscopic chemical reactions, e.g. . The thing is, there was still the possibility that those proportions were made up of something continuous that for some reason could only combine in the given proportions, so the atoms could only be strictly consider calculatory devices pending further evidence.
Subtle is the Lord by Abraham Pais (1982) chapter 5 "The reality of molecules" has some good mentions. Notably, physicists generally came to believe in atoms earlier than chemists, because the phenomena they were most interested in, e.g. pressure in the ideal gas law, and then Maxwell-Boltzmann statistics just scream atoms more loudly than chemical reactions, as they saw that these phenomena could be explained to some degree by traditional mechanics of little balls.
Confusion around the probabilistic nature of the second law of thermodynamics was also used as a physical counterargument by some. Pais mentions that Wilhelm Ostwald notably argued that the time reversibility of classical mechanics + the second law being a fundamental law of physics (and not just probabilistic, which is the correct hypothesis as we now understand) must imply that atoms are not classic billiard balls, otherwise the second law could be broken.
Pais also mentions that a big "chemical" breakthrough was isomers suggest that atoms exist.
Very direct evidence evidence:
- Brownian motion mathematical analysis in 1908. Brownian motion just makes it too clear that liquids cannot be continuous... if they were, there would obviously be no Brownian motion, full stop.
- X-ray crystallography: it sees crystal latices

Figure 1. Still from A boy and his atom by IBM. Source.
Less direct evidence:
- 1874 Isomers suggest that atoms exist
- kinetic theory of gases seems to explain certain phenomena really well
Subtle is the Lord by Abraham Pais (1982) page 40 mentions several methods that Einstein used to "prove" that atoms were real. Perhaps the greatest argument of all is that several unrelated methods give the same estimates of atom size/mass:
- from 1905:
- in light quantum paper
- enabled by experimental work of Wilhelm Pfeffer on producing rigid membranes
- 1911: blueness of the sky and critical opalescence
Benzene Created 2024-07-04 Updated 2025-07-16

.jpg){kind=link}
{kind=link}
Cryogenic electron microscopy Updated 2025-07-16
This technique has managed to determine protein 3D structures for proteins that people were not able to crystallize for X-ray crystallography.
It is said however that cryoEM is even fiddlier than X-ray crystallography, so it is mostly attempted if crystallization attempts fail.
We just put a gazillion copies of our molecule of interest in a solution, and then image all of them in the frozen water.
Each one of them appears in the image in a random rotated view, so given enough of those point of view images, we can deduce the entire 3D structure of the molecule.
Ciro Santilli once watched a talk by Richard Henderson about cryoEM circa 2020, where he mentioned that he witnessed some students in the 1980's going to Germany, and coming into contact with early cryoEM. And when they came back, they just told their principal investigator: "I'm going to drop my PhD theme and focus exclusively on cryoEM". That's how hot the cryo thing was! So cool.

{kind=link}
E. Coli K-12 MG1655 gene thrA Updated 2025-07-16
NCBI entry: www.ncbi.nlm.nih.gov/gene/945803.
This protein is an enzyme. The UniProt entry clearly shows the chemical reactions that it catalyses. In this case, there are actually two! It can either transforming the metabolite:Also interestingly, we see that both of those reaction require some extra energy to catalyse, one needing adenosine triphosphate and the other nADP+.
TODO: any mention of how much faster it makes the reaction, numerically?
Since this is an enzyme, it would also be interesting to have a quick search for it in the KEGG entry starting from the organism: www.genome.jp/pathway/eco01100+M00022 We type in the search bar "thrA", it gives a long list, but the last entry is our "thrA". Selecting it highlights two pathways in the large graph, so we understand that it catalyzes two different reactions, as suggested by the protein name itself (fused blah blah). We can now hover over:Note that common cofactor are omitted, since we've learnt from the UniProt entry that this reaction uses ATP.
- the edge: it shows all the enzymes that catalyze the given reaction. Both edges actually have multiple enzymes, e.g. the L-Homoserine path is also catalyzed by another enzyme called metL.
- the node: they are the metabolites, e.g. one of the paths contains "L-homoserine" on one node and "L-aspartate 4-semialdehyde"
If we can now click on the L-Homoserine edge, it takes us to: www.genome.jp/entry/eco:b0002+eco:b3940. Under "Pathway" we see an interesting looking pathway "Glycine, serine and threonine metabolism": www.genome.jp/pathway/eco00260+b0002 which contains a small manually selected and extremely clearly named subset of the larger graph!
But looking at the bottom of this subgraph (the UI is not great, can't Ctrl+F and enzyme names not shown, but the selected enzyme is slightly highlighted in red because it is in the URL www.genome.jp/pathway/eco00260+b0002 vs www.genome.jp/pathway/eco00260) we clearly see that thrA, thrB and thrC for a sequence that directly transforms "L-aspartate 4-semialdehyde" into "Homoserine" to "O-Phospho-L-homoserine" and finally tothreonine. This makes it crystal clear that they are not just located adjacently in the genome by chance: they are actually functionally related, and likely controlled by the same transcription factor: when you want one of them, you basically always want the three, because you must be are lacking threonine. TODO find transcription factor!
The UniProt entry also shows an interactive browser of the tertiary structure of the protein. We note that there are currently two sources available: X-ray crystallography and AlphaFold. To be honest, the AlphaFold one looks quite off!!!
By inspecting the FASTA for the entire genome, or by using the NCBI open reading frame tool, we see that this gene lies entirely in its own open reading frame, so it is quite boring
From the FASTA we see that the very first three Codons at position 337 arewhere
ATG CGA GTGATG is the start codon, and CGA GTG should be the first two that actually go into the protein:ecocyc.org/gene?orgid=ECOLI&id=ASPKINIHOMOSERDEHYDROGI-MONOMER mentions that the enzime is most active as protein complex with four copies of the same protein:TODO image?
Aspartate kinase I / homoserine dehydrogenase I comprises a dimer of ThrA dimers. Although the dimeric form is catalytically active, the binding equilibrium dramatically favors the tetrameric form. The aspartate kinase and homoserine dehydrogenase activities of each ThrA monomer are catalyzed by independent domains connected by a linker region.
Electron crystallography Updated 2025-07-16
Crystallography determination with a transmission electron microscopy instead of the more classical X-ray crystallography.
Lysozyme Updated 2025-07-16
The second protein to have its structure determined, after myoglobin, by X-ray crystallography, in 1965.
Breaks up peptidoglycan present in the bacterial cell wall, which is thicker in Gram-positive bacteria, which is what this enzyme seems to target.
Part of the inate immune system.
Lysozyme structure resolution (1965) Updated 2025-07-16
With X-ray crystallography by David Chilton Phillips. The second protein to be resolved fter after myoglobin, and the first enzyme.
Published at: Structure of Hen Egg-White Lysozyme: A Three-dimensional Fourier Synthesis at 2 Å Resolution (1965). The work was done while at the Davy Faraday Research Laboratory of the Royal Institution.
Phillips also published a lower resolution (6angstrom) of the enzyme-inhibitor complexes at about the same time: Structure of Some Crystalline Lysozyme-Inhibitor Complexes Determined by X-Ray Analysis At 6 Å Resolution (1965). The point of doing this is that it points out the active site of the enzyme.
Optical microscope Updated 2025-07-16
Quantum chemistry Updated 2025-07-16
Ah, the jewel of computational physics.
Also known as an ab initio method: no experimental measurement is taken as input, QED is all you need.
But since QED is thought to fully describe all relevant aspects molecules, it could be called "the" ab initio method.
For one, if we were able to predict protein molecule interactions, our understanding of molecular biology technologies would be solved.
No more ultra expensive and complicated X-ray crystallography or cryogenic electron microscopy.
And the fact that quantum computers are one of the most promising advances to this field, is also very very exciting: Section "Quantum algorithm".
Synchrotron Updated 2025-07-16
Most important application: produce X-rays for X-ray crystallography.
Note however that the big experiments at CERN, like the Large Hadron Collider, are also synchrotrons.
List of facilities: en.wikipedia.org/wiki/List_of_synchrotron_radiation_facilities
X-ray diffraction Updated 2025-07-16
Often used as a synonym for X-ray crystallography, or to refer more specifically to the diffraction part of the experiment (exluding therefore sample preparation and data processing).