
Incoming links: Chemical reaction
E. Coli K-12 MG1655 gene thrA Updated 2025-07-16
NCBI entry: www.ncbi.nlm.nih.gov/gene/945803.
This protein is an enzyme. The UniProt entry clearly shows the chemical reactions that it catalyses. In this case, there are actually two! It can either transforming the metabolite:Also interestingly, we see that both of those reaction require some extra energy to catalyse, one needing adenosine triphosphate and the other nADP+.
TODO: any mention of how much faster it makes the reaction, numerically?
Since this is an enzyme, it would also be interesting to have a quick search for it in the KEGG entry starting from the organism: www.genome.jp/pathway/eco01100+M00022 We type in the search bar "thrA", it gives a long list, but the last entry is our "thrA". Selecting it highlights two pathways in the large graph, so we understand that it catalyzes two different reactions, as suggested by the protein name itself (fused blah blah). We can now hover over:Note that common cofactor are omitted, since we've learnt from the UniProt entry that this reaction uses ATP.
- the edge: it shows all the enzymes that catalyze the given reaction. Both edges actually have multiple enzymes, e.g. the L-Homoserine path is also catalyzed by another enzyme called metL.
- the node: they are the metabolites, e.g. one of the paths contains "L-homoserine" on one node and "L-aspartate 4-semialdehyde"
If we can now click on the L-Homoserine edge, it takes us to: www.genome.jp/entry/eco:b0002+eco:b3940. Under "Pathway" we see an interesting looking pathway "Glycine, serine and threonine metabolism": www.genome.jp/pathway/eco00260+b0002 which contains a small manually selected and extremely clearly named subset of the larger graph!
But looking at the bottom of this subgraph (the UI is not great, can't Ctrl+F and enzyme names not shown, but the selected enzyme is slightly highlighted in red because it is in the URL www.genome.jp/pathway/eco00260+b0002 vs www.genome.jp/pathway/eco00260) we clearly see that thrA, thrB and thrC for a sequence that directly transforms "L-aspartate 4-semialdehyde" into "Homoserine" to "O-Phospho-L-homoserine" and finally tothreonine. This makes it crystal clear that they are not just located adjacently in the genome by chance: they are actually functionally related, and likely controlled by the same transcription factor: when you want one of them, you basically always want the three, because you must be are lacking threonine. TODO find transcription factor!
The UniProt entry also shows an interactive browser of the tertiary structure of the protein. We note that there are currently two sources available: X-ray crystallography and AlphaFold. To be honest, the AlphaFold one looks quite off!!!
By inspecting the FASTA for the entire genome, or by using the NCBI open reading frame tool, we see that this gene lies entirely in its own open reading frame, so it is quite boring
From the FASTA we see that the very first three Codons at position 337 arewhere
ATG CGA GTGATG is the start codon, and CGA GTG should be the first two that actually go into the protein:ecocyc.org/gene?orgid=ECOLI&id=ASPKINIHOMOSERDEHYDROGI-MONOMER mentions that the enzime is most active as protein complex with four copies of the same protein:TODO image?
Aspartate kinase I / homoserine dehydrogenase I comprises a dimer of ThrA dimers. Although the dimeric form is catalytically active, the binding equilibrium dramatically favors the tetrameric form. The aspartate kinase and homoserine dehydrogenase activities of each ThrA monomer are catalyzed by independent domains connected by a linker region.
E. Coli Whole Cell Model by Covert Lab Source code overview Updated 2025-07-16
Let's try to understand some interesting looking, with a special focus on our understanding of the tiny E. Coli K-12 MG1655 operon thrLABC part of the metabolism, which we have well understood at Section "E. Coli K-12 MG1655 operon thrLABC".
reconstruction/ecoli/flat/compartments.tsvcontains cellular compartment information:"abbrev" "id" "n" "CCO-BAC-NUCLEOID" "j" "CCO-CELL-PROJECTION" "w" "CCO-CW-BAC-NEG" "c" "CCO-CYTOSOL" "e" "CCO-EXTRACELLULAR" "m" "CCO-MEMBRANE" "o" "CCO-OUTER-MEM" "p" "CCO-PERI-BAC" "l" "CCO-PILUS" "i" "CCO-PM-BAC-NEG"CCO: "Celular COmpartment"BAC-NUCLEOID: nucleoidCELL-PROJECTION: cell projectionCW-BAC-NEG: TODO confirm: cell wall (of a Gram-negative bacteria)CYTOSOL: cytosolEXTRACELLULAR: outside the cellMEMBRANE: cell membraneOUTER-MEM: bacterial outer membranePERI-BAC: periplasmPILUS: pilusPM-BAC-NEG: TODO: plasma membrane, but that is the same as cell membrane no?
reconstruction/ecoli/flat/promoters.tsvcontains promoter information. Simple file, sample lines:corresponds to E. Coli K-12 MG1655 promoter thrLp, which starts as position 148."position" "direction" "id" "name" 148 "+" "PM00249" "thrLp"reconstruction/ecoli/flat/proteins.tsvcontains protein information. Sample line corresponding to e. Coli K-12 MG1655 gene thrA:so we understand that:"aaCount" "name" "seq" "comments" "codingRnaSeq" "mw" "location" "rnaId" "id" "geneId" [91, 46, 38, 44, 12, 53, 30, 63, 14, 46, 89, 34, 23, 30, 29, 51, 34, 4, 20, 0, 69] "ThrA" "MRVL..." "Location information from Ecocyc dump." "AUGCGAGUGUUG..." [0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 89103.51099999998, 0.0, 0.0, 0.0, 0.0] ["c"] "EG10998_RNA" "ASPKINIHOMOSERDEHYDROGI-MONOMER" "EG10998"aaCount: amino acid count, how many of each of the 20 proteinogenic amino acid are thereseq: full sequence, using the single letter abbreviation of the proteinogenic amino acidsmw; molecular weight? The 11 components appear to be given atreconstruction/ecoli/flat/scripts/unifyBulkFiles.py:so they simply classify the weight? Presumably this exists for complexes that have multiple classes?molecular_weight_keys = [ '23srRNA', '16srRNA', '5srRNA', 'tRNA', 'mRNA', 'miscRNA', 'protein', 'metabolite', 'water', 'DNA', 'RNA' # nonspecific RNA ]23srRNA,16srRNA,5srRNAare the three structural RNAs present in the ribosome: 23S ribosomal RNA, 16S ribosomal RNA, 5S ribosomal RNA, all others are obvious:- tRNA
- mRNA
- protein. This is the seventh class, and this enzyme only contains mass in this class as expected.
- metabolite
- water
- DNA
- RNA: TODO
rnavsmiscRNA
location: cell compartment where the protein is present,cdefined atreconstruction/ecoli/flat/compartments.tsvas cytoplasm, as expected for something that will make an amino acid
reconstruction/ecoli/flat/rnas.tsv: TODO vstranscriptionUnits.tsv. Sample lines:"halfLife" "name" "seq" "type" "modifiedForms" "monomerId" "comments" "mw" "location" "ntCount" "id" "geneId" "microarray expression" 174.0 "ThrA [RNA]" "AUGCGAGUGUUG..." "mRNA" [] "ASPKINIHOMOSERDEHYDROGI-MONOMER" "" [0.0, 0.0, 0.0, 0.0, 790935.00399999996, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0] ["c"] [553, 615, 692, 603] "EG10998_RNA" "EG10998" 0.0005264904halfLife: half-lifemw: molecular weight, same as inreconstruction/ecoli/flat/proteins.tsv. This molecule only have weight in themRNAclass, as expected, as it just codes for a proteinlocation: same as inreconstruction/ecoli/flat/proteins.tsvntCount: nucleotide count for each of the ATGCmicroarray expression: presumably refers to DNA microarray for gene expression profiling, but what measure exactly?
reconstruction/ecoli/flat/sequence.fasta: FASTA DNA sequence, first two lines:>E. coli K-12 MG1655 U00096.2 (1 to 4639675 = 4639675 bp) AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGreconstruction/ecoli/flat/transcriptionUnits.tsv: transcription units. We can observe for example the two different transcription units of the E. Coli K-12 MG1655 operon thrLABC in the lines:"expression_rate" "direction" "right" "terminator_id" "name" "promoter_id" "degradation_rate" "id" "gene_id" "left" 0.0 "f" 310 ["TERM0-1059"] "thrL" "PM00249" 0.198905992329492 "TU0-42486" ["EG11277"] 148 657.057317358791 "f" 5022 ["TERM_WC-2174"] "thrLABC" "PM00249" 0.231049060186648 "TU00178" ["EG10998", "EG10999", "EG11000", "EG11277"] 148promoter_id: matches promoter id inreconstruction/ecoli/flat/promoters.tsvgene_id: matches id inreconstruction/ecoli/flat/genes.tsvid: matches exactly those used in BioCyc, which is quite nice, might be more or less standardized:
reconstruction/ecoli/flat/genes.tsv"length" "name" "seq" "rnaId" "coordinate" "direction" "symbol" "type" "id" "monomerId" 66 "thr operon leader peptide" "ATGAAACGCATT..." "EG11277_RNA" 189 "+" "thrL" "mRNA" "EG11277" "EG11277-MONOMER" 2463 "ThrA" "ATGCGAGTGTTG" "EG10998_RNA" 336 "+" "thrA" "mRNA" "EG10998" "ASPKINIHOMOSERDEHYDROGI-MONOMER"reconstruction/ecoli/flat/metabolites.tsvcontains metabolite information. Sample lines:In the case of the enzyme thrA, one of the two reactions it catalyzes is "L-aspartate 4-semialdehyde" into "Homoserine"."id" "mw7.2" "location" "HOMO-SER" 119.12 ["n", "j", "w", "c", "e", "m", "o", "p", "l", "i"] "L-ASPARTATE-SEMIALDEHYDE" 117.104 ["n", "j", "w", "c", "e", "m", "o", "p", "l", "i"]Starting from the enzyme page: biocyc.org/gene?orgid=ECOLI&id=EG10998 we reach the reaction page: biocyc.org/ECOLI/NEW-IMAGE?type=REACTION&object=HOMOSERDEHYDROG-RXN which has reaction IDHOMOSERDEHYDROG-RXN, and that page which clarifies the IDs:so these are the compounds that we care about.- biocyc.org/compound?orgid=ECOLI&id=L-ASPARTATE-SEMIALDEHYDE: "L-aspartate 4-semialdehyde" has ID
L-ASPARTATE-SEMIALDEHYDE - biocyc.org/compound?orgid=ECOLI&id=HOMO-SER: "Homoserine" has ID
HOMO-SER
- biocyc.org/compound?orgid=ECOLI&id=L-ASPARTATE-SEMIALDEHYDE: "L-aspartate 4-semialdehyde" has ID
reconstruction/ecoli/flat/reactions.tsvcontains chemical reaction information. Sample lines:"reaction id" "stoichiometry" "is reversible" "catalyzed by" "HOMOSERDEHYDROG-RXN-HOMO-SER/NAD//L-ASPARTATE-SEMIALDEHYDE/NADH/PROTON.51." {"NADH[c]": -1, "PROTON[c]": -1, "HOMO-SER[c]": 1, "L-ASPARTATE-SEMIALDEHYDE[c]": -1, "NAD[c]": 1} false ["ASPKINIIHOMOSERDEHYDROGII-CPLX", "ASPKINIHOMOSERDEHYDROGI-CPLX"] "HOMOSERDEHYDROG-RXN-HOMO-SER/NADP//L-ASPARTATE-SEMIALDEHYDE/NADPH/PROTON.53." {"NADPH[c]": -1, "NADP[c]": 1, "PROTON[c]": -1, "L-ASPARTATE-SEMIALDEHYDE[c]": -1, "HOMO-SER[c]": 1 false ["ASPKINIIHOMOSERDEHYDROGII-CPLX", "ASPKINIHOMOSERDEHYDROGI-CPLX"]catalized by: here we seeASPKINIHOMOSERDEHYDROGI-CPLX, which we can guess is a protein complex made out ofASPKINIHOMOSERDEHYDROGI-MONOMER, which is the ID for thethrAwe care about! This is confirmed incomplexationReactions.tsv.
reconstruction/ecoli/flat/complexationReactions.tsvcontains information about chemical reactions that produce protein complexes:The"process" "stoichiometry" "id" "dir" "complexation" [ { "molecule": "ASPKINIHOMOSERDEHYDROGI-CPLX", "coeff": 1, "type": "proteincomplex", "location": "c", "form": "mature" }, { "molecule": "ASPKINIHOMOSERDEHYDROGI-MONOMER", "coeff": -4, "type": "proteinmonomer", "location": "c", "form": "mature" } ] "ASPKINIHOMOSERDEHYDROGI-CPLX_RXN" 1coeffis how many monomers need to get together for form the final complex. This can be seen from the Summary section of ecocyc.org/gene?orgid=ECOLI&id=ASPKINIHOMOSERDEHYDROGI-MONOMER:Fantastic literature summary! Can't find that in database form there however.Aspartate kinase I / homoserine dehydrogenase I comprises a dimer of ThrA dimers. Although the dimeric form is catalytically active, the binding equilibrium dramatically favors the tetrameric form. The aspartate kinase and homoserine dehydrogenase activities of each ThrA monomer are catalyzed by independent domains connected by a linker region.
reconstruction/ecoli/flat/proteinComplexes.tsvcontains protein complex information:"name" "comments" "mw" "location" "reactionId" "id" "aspartate kinase / homoserine dehydrogenase" "" [0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 356414.04399999994, 0.0, 0.0, 0.0, 0.0] ["c"] "ASPKINIHOMOSERDEHYDROGI-CPLX_RXN" "ASPKINIHOMOSERDEHYDROGI-CPLX"reconstruction/ecoli/flat/protein_half_lives.tsvcontains the half-life of proteins. Very few proteins are listed however for some reason.reconstruction/ecoli/flat/tfIds.csv: transcription factors information:"TF" "geneId" "oneComponentId" "twoComponentId" "nonMetaboliteBindingId" "activeId" "notes" "arcA" "EG10061" "PHOSPHO-ARCA" "PHOSPHO-ARCA" "fnr" "EG10325" "FNR-4FE-4S-CPLX" "FNR-4FE-4S-CPLX" "dksA" "EG10230"
Enzyme Updated 2025-07-16
For an initial concrete example, consider e. Coli K-12 MG1655 gene thrA.
How Enzymes Work by RCSBProteinDataBank (2017)
Source. Shows in detail how aconitase catalyses the citrate to isocitrate reaction in the citric acid cycle. Maxwell-Boltzmann vs Bose-Einstein vs Fermi-Dirac statistics Created 2024-10-28 Updated 2025-07-16
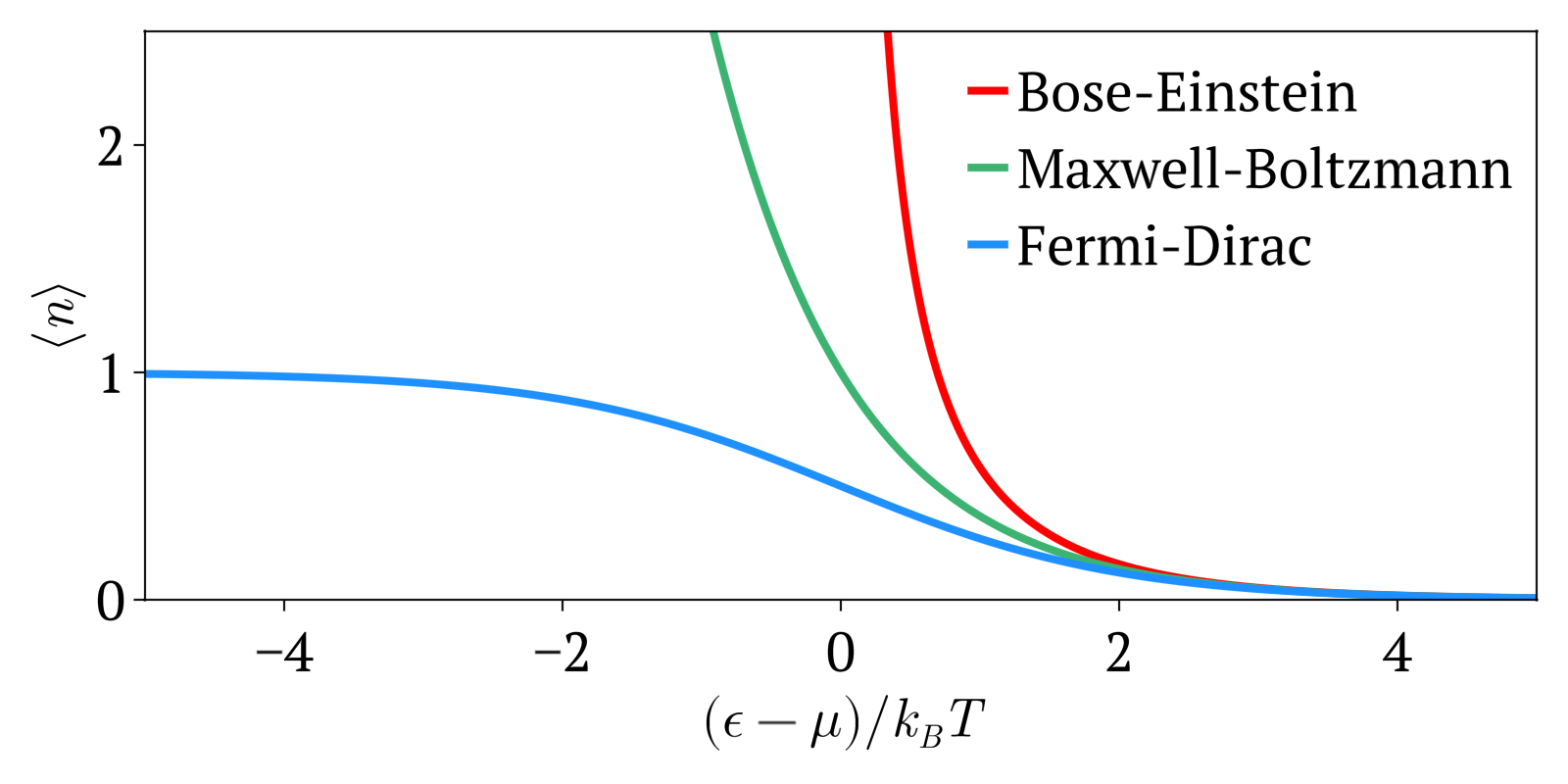
Maxwell-Boltzmann statistics, Bose-Einstein statistics and Fermi-Dirac statistics all describe how energy is distributed in different physical systems at a given temperature.
For example, Maxwell-Boltzmann statistics describes how the speeds of particles are distributed in an ideal gas.
The temperature of a gas is only a statistical average of the total energy of the gas. But at a given temperature, not all particles have the exact same speed as the average: some are higher and others lower than the average.
For a large number of particles however, the fraction of particles that will have a given speed at a given temperature is highly deterministic, and it is this that the distributions determine.
One of the main interest of learning those statistics is determining the probability, and therefore average speed, at which some event that requires a minimum energy to happen happens. For example, for a chemical reaction to happen, both input molecules need a certain speed to overcome the potential barrier of the reaction. Therefore, if we know how many particles have energy above some threshold, then we can estimate the speed of the reaction at a given temperature.
The three distributions can be summarized as:
- Maxwell-Boltzmann statistics: statistics without considering quantum statistics. It is therefore only an approximation. The other two statistics are the more precise quantum versions of Maxwell-Boltzmann and tend to it at high temperatures or low concentration. Therefore this one works well at high temperatures or low concentrations.
- Bose-Einstein statistics: quantum version of Maxwell-Boltzmann statistics for bosons
- Fermi-Dirac statistics: quantum version of Maxwell-Boltzmann statistics for fermions. Sample system: electrons in a metal, which creates the free electron model. Compared to Maxwell-Boltzmann statistics, this explained many important experimental observations such as the specific heat capacity of metals. A very cool and concrete example can be seen at youtu.be/5V8VCFkAd0A?t=1187 from Video "Using a Photomultiplier to Detect single photons by Huygens Optics" where spontaneous field electron emission would follow Fermi-Dirac statistics. In this case, the electrons with enough energy are undesired and a source of noise in the experiment.
A good conceptual starting point is to like the example that is mentioned at The Harvest of a Century by Siegmund Brandt (2008).
Consider a system with 2 particles and 3 states. Remember that:
- in quantum statistics (Bose-Einstein statistics and Fermi-Dirac statistics), particles are indistinguishable, therefore, we might was well call both of them
A, as opposed toAandBfrom non-quantum statistics - in Bose-Einstein statistics, two particles may occupy the same state. In Fermi-Dirac statistics
Therefore, all the possible way to put those two particles in three states are for:
- Maxwell-Boltzmann distribution: both A and B can go anywhere:
- Bose-Einstein statistics: because A and B are indistinguishable, there is now only 1 possibility for the states where A and B would be in different states.
- Fermi-Dirac statistics: now states with two particles in the same state are not possible anymore:
Molecular biology technologies Updated 2025-07-16
As of 2019, the silicon industry is ending, and molecular biology technology is one of the most promising and growing field of engineering.

Ciro Santilli is especially excited about DNA-related technologies, because DNA is the centerpiece of biology, and it is programmable.
First, during the 2000's, the cost of DNA sequencing fell to about 1000 USD per genome in the end of the 2010's: Figure 2. "Cost per genome vs Moore's law from 2000 to 2019", largely due to "Illumina's" technology.
The medical consequences of this revolution are still trickling down towards medical applications of 2019, inevitably, but somewhat slowly due to tight privacy control of medical records.

{kind=link}
{kind=link}
Ciro Santilli predicts that when the 100 dollar mark is reached, every person of the First world will have their genome sequenced, and then medical applications will be closer at hand than ever.
But even 100 dollars is not enough. Sequencing power is like computing power: humankind can never have enough. Sequencing is not a one per person thing. For example, as of 2019 tumors are already being sequenced to help understand and treat them, and scientists/doctors will sequence as many tumor cells as budget allows.
Then, in the 2010's, CRISPR/Cas9 gene editing started opening up the way to actually modifying the genome that we could now see through sequencing.
What's next?
Ciro believes that the next step in the revolution could be could be: de novo DNA synthesis.
This technology could be the key to the one of the ultimate dream of biologists: cheap programmable biology with push-button organism bootstrap!
Just imagine this: at the comfort of your own garage, you take some model organism of interest, maybe start humble with Escherichia coli. Then you modify its DNA to your liking, and upload it to a 3D printer sized machine on your workbench, which automatically synthesizes the DNA, and injects into a bootstrapped cell.
You then make experiments to check if the modified cell achieves your desired new properties, e.g. production of some protein, and if not reiterate, just like a software engineer.
Of course, even if we were able to do the bootstrap, the debugging process then becomes key, as visibility is the key limitation of biology, maybe we need other cheap technologies to come in at that point.
This a place point we see the beauty of evolution the brightest: evolution does not require observability. But it also implies that if your changes to the organism make it less fit, then your mutation will also likely be lost. This has to be one of the considerations done when designing your organism.
Other cool topic include:
- computational biology: simulations of cell metabolism, protein and small molecule, including computational protein folding and chemical reactions. This is basically the simulation part of omics.If we could only simulate those, we would basically "solve molecular biology". Just imagine, instead of experimenting for a hole year, the 2021 Nobel Prize in Physiology and Medicine could have been won from a few hours on a supercomputer to determine which protein had the desired properties, using just DNA sequencing as a starting point!
- microscopy: crystallography, cryoEM
- analytical chemistry: mass spectroscopy, single cell analysis (Single-cell RNA sequencing)
Ciro is sad that by the time he dies, humanity won't have understood the human brain, maybe not even a measly Escherichia coli... Heck, even key molecular biology events are not yet fully understood, see e.g. transcription regulation.
One of the most exciting aspects of molecular biology technologies is their relatively low entry cost, compared for example to other areas such as fusion energy and quantum computing.